Hemocromatosis.
Reading File
Finding Sources
Searching PubMed
hemochromatosis
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Loading Image
I now have everything needed for a comprehensive response. Here is the full clinical overview of hemochromatosis:
Hemocromatosis (Hemochromatosis)
Definición y Clasificación
La hemocromatosis es un trastorno caracterizado por la absorción intestinal excesiva de hierro, con depósito progresivo en órganos como hígado, páncreas, corazón, articulaciones y órganos endocrinos. La acumulación normal de hierro en adultos es de 3-4 g (aproximadamente 0,5 g almacenado en hepatocitos); en hemocromatosis severa, el hierro total puede superar los 50 g, acumulándose más de un tercio en el hígado.
Clasificación de la sobrecarga de hierro:
| Tipo | Causas |
|---|---|
| Hemocromatosis Hereditaria | Mutaciones en HFE, TFR2, ferroportin (adultos); HJV, HAMP (juvenil) |
| Hemosiderosis Secundaria | Transfusiones repetidas, hemólisis crónica, talasemia grave, aplasia medular |
| Asociada a hematopoyesis ineficaz | β-talasemia, síndrome mielodisplásico |
| Mayor ingesta oral | Siderosis africana (Bantu) |
| Hepatopatía crónica | Enfermedad hepática alcohólica, porfiria cutánea tarda |
| Neonatal | Hemocromatosis neonatal (no hereditaria, desarrollada in utero) |
- Robbins & Kumar Basic Pathology, p. 613 | Robbins, Cotran & Kumar, p. 787
Patogénesis
El regulador central de la absorción intestinal de hierro es la hepcidina (proteína codificada por el gen HAMP, producida por el hígado).
Mecanismo normal:
- HFE, HJV y TFR2 (expresados en hepatocitos) actúan como "sensores" de hierro
- Cuando el hierro es abundante, estas proteínas estimulan la síntesis de hepcidina
- La hepcidina circula hacia el intestino y se une a la ferroportina en enterocitos → internalización y degradación de ferroportina → reduce el eflujo de hierro desde enterocitos
En hemocromatosis hereditaria:
- Mutaciones de pérdida de función en HFE, HJV, TFR2 o HAMP → síntesis disminuida de hepcidina → ferroportina no se degrada → aumento del eflujo de hierro → sobrecarga sistémica
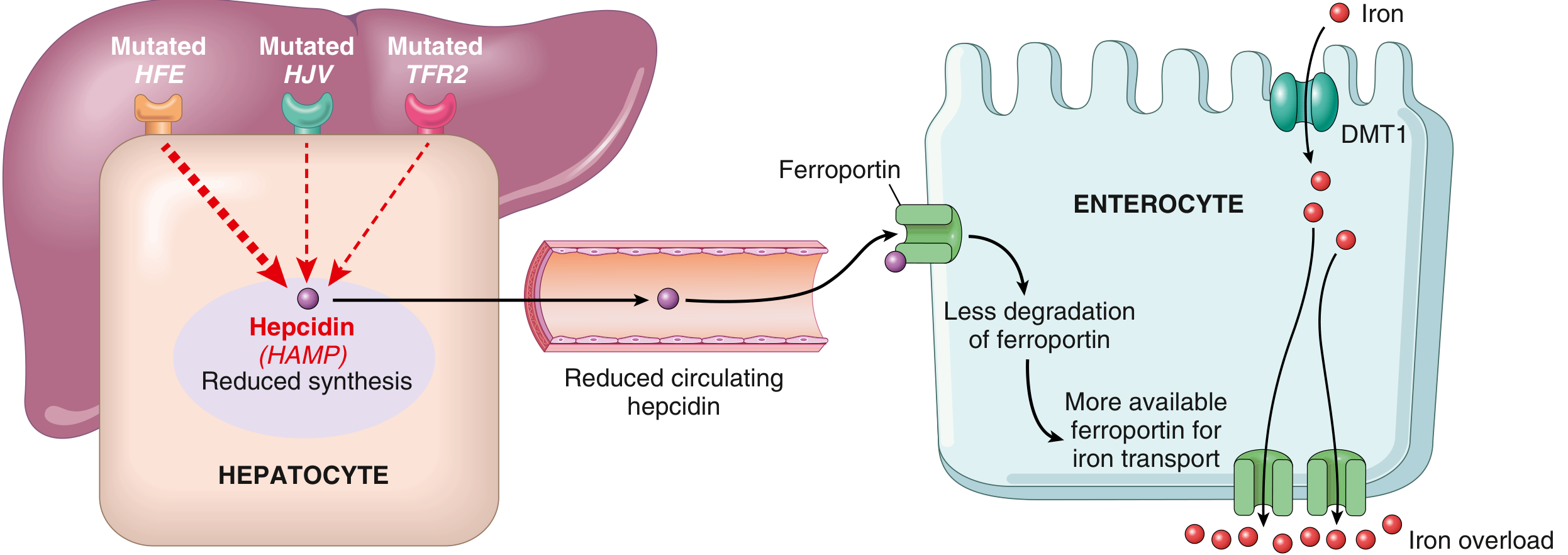
FIG. Patogénesis: En hemocromatosis hereditaria, las mutaciones en HFE, HJV o TFR2 reducen la síntesis de hepcidina. Con menos hepcidina circulante, la ferroportina no se degrada, hay mayor transporte de hierro desde los enterocitos y sobrecarga sistémica. - Robbins & Kumar Basic Pathology
Hemocromatosis secundaria: La eritropoyesis ineficaz crónica (ej. β-talasemia) eleva la eritroferrina (hormona de eritroblastos medulares), que suprime la hepcidina, con el mismo resultado final.
Mecanismos de daño tisular
- Peroxidación lipídica por reacciones de radicales libres catalizadas por hierro
- Estimulación de células estrelladas hepáticas → formación de colágeno → fibrosis
- Daño al ADN por ROS → lesión celular letal o predisposición a carcinoma hepatocelular
- Robbins & Kumar Basic Pathology, p. 614
Genética
Forma adulta (la más común):
- Gen HFE mutado: codifica una molécula similar a HLA clase I que regula la síntesis de hepcidina
- Mutación C282Y (sustitución cisteína→tirosina en posición 282): presente en >70% de los pacientes con HH. Es casi exclusiva de poblaciones de origen europeo
- Mutación H63D (histidina→aspartato en posición 63): distribución mundial, menos penetrante
- Frecuencia de homocigosidad C282Y en europeos: ~0,45% (1 en 220 personas), pero solo ~10% desarrollan enfermedad clínica
Formas menos comunes:
-
Mutaciones en TFR2 (receptor de transferrina 2) → hemocromatosis adulta autosómica recesiva
-
Mutaciones en SLC40A1 (ferroportina) → hemocromatosis adulta autosómica dominante
-
Mutaciones en HAMP o HJV (hemojuvelina) → hemocromatosis juvenil (formas autosómicas recesivas, de inicio temprano y más severas)
-
Andrews' Diseases of the Skin, p. 999 | Robbins, Cotran & Kumar, p. 788
Características Clínicas
Los síntomas suelen aparecer en la 4.ª-6.ª décadas en hombres y más tarde en mujeres (la menstruación contrarresta la acumulación hasta la menopausia). La proporción hombre:mujer con sobrecarga clínicamente significativa es de 5-7:1.
Tríada clásica (en casos plenamente desarrollados):
- Cirrosis micronodular - 100% de los casos graves
- Diabetes mellitus - 75-80% de los pacientes (destrucción de islotes pancreáticos)
- Hiperpigmentación cutánea - 75-80% de los pacientes
Otras manifestaciones:
- Hepáticas: hepatomegalia, dolor abdominal, alteración de PFH → cirrosis → carcinoma hepatocelular (riesgo aumentado 200 veces en enfermedad no tratada)
- Cardíacas: arritmias, miocardiopatía, insuficiencia cardíaca
- Articulares: artropatía atípica (~40-50% de los pacientes), pseudogota por depósito de pirofosfato cálcico
- Endocrinas: hipogonadismo (amenorrea en la mujer, impotencia y pérdida de libido en el hombre), hipopituitarismo
- Cutáneas: ver sección aparte
Factores que aceleran el desarrollo clínico: consumo de alcohol, tabaquismo, coinfección con VHC.
- Robbins & Kumar Basic Pathology, p. 615 | Andrews' Diseases of the Skin, p. 999
Manifestaciones Cutáneas
La pigmentación mucocutánea gris-marrón es la manifestación cutánea característica. Se acentúa en áreas fotoexpuestas (antebrazos, dorso de manos, cara) y en el área inguinal.
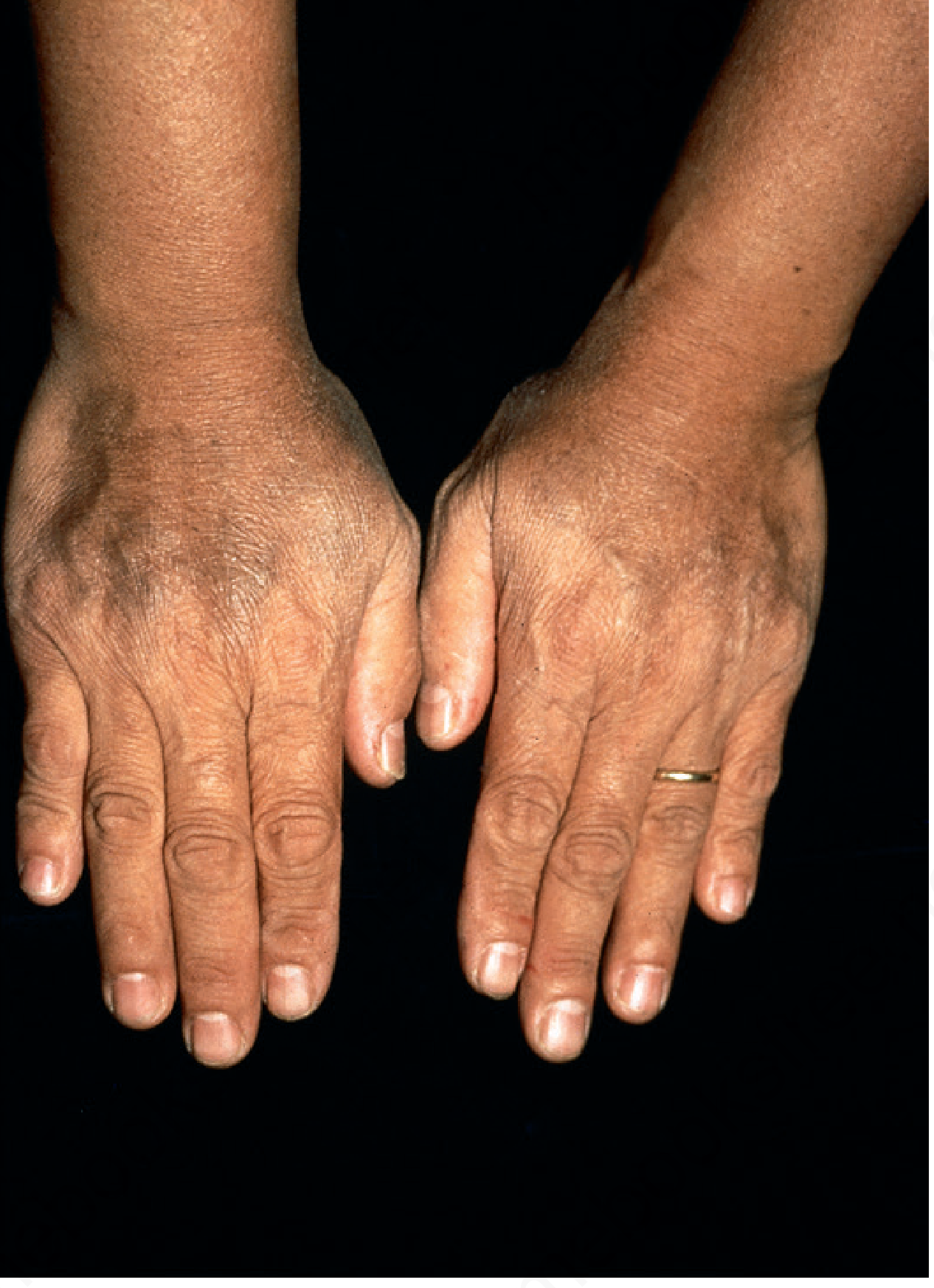
Fig. Hemocromatosis: hiperpigmentación gris-parduzca en el dorso de las manos - Andrews' Diseases of the Skin
Mecanismo de la pigmentación: aunque hay depósito dérmico de hemosiderina en macrófagos y fibroblastos, la mayor parte de la coloración visible se debe a aumento de melanina epidérmica en la capa basal. La combinación produce un tinte grisáceo característico.
Otras manifestaciones cutáneas:
- Mucosas pigmentadas (hasta 20% de los pacientes)
- Koiloniquia
- Ictiosis localizada
- Alopecia (frecuente)
- Prurito
- Porfiria cutánea tarda (más frecuente en hemocromatosis por inhibición de la uroporfirinógeno descarboxilasa hepática por sobrecarga de hierro)
- Mayor riesgo de úlceras en piernas en pacientes con insuficiencia venosa crónica y mutación C282Y (riesgo aumentado 6 veces)
Prevalencia de pigmentación: ~30% en hombres afectados, <10% en mujeres diagnosticadas.
- Andrews' Diseases of the Skin, p. 999
Morfología Patológica
Depósito de hemosiderina (orden decreciente de severidad):
Hígado > Páncreas > Miocardio > Hipófisis > Suprarrenales > Tiroides/Paratiroides > Articulaciones > Piel
Hígado:
- Inicialmente: gránulos dorados de hemosiderina en citoplasma de hepatocitos periportales (teñibles con Prussian blue)
- Progresión: depósito en todo el lóbulo, epitelio biliar y células de Kupffer
- Hígado levemente aumentado, color chocolate
- Fibrosis → cirrosis micronodular con pigmentación intensa (marrón muy oscuro a negro)
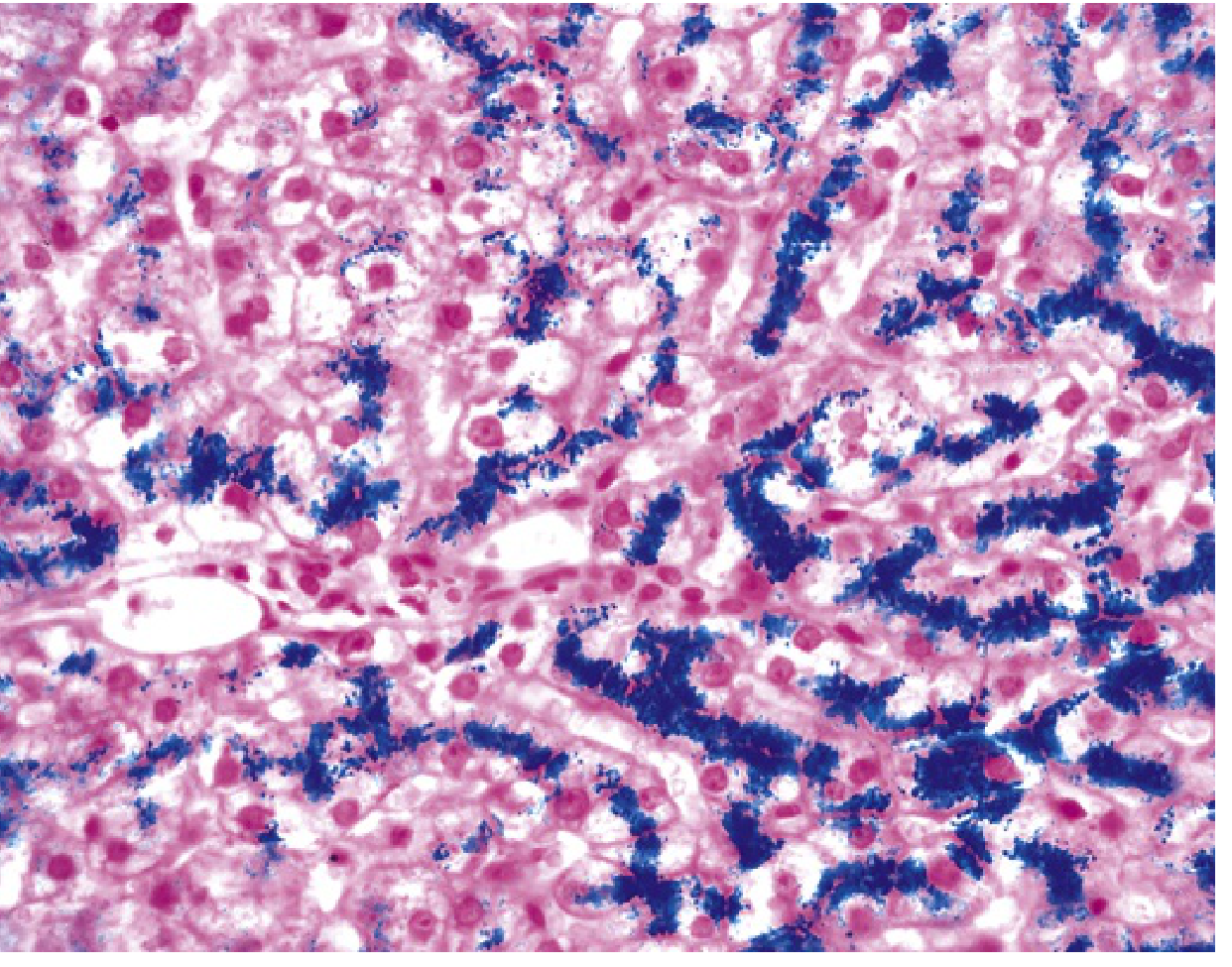
FIG. Hemocromatosis hereditaria. Tinción de Prussian blue: el hierro hepatocelular aparece azul. La arquitectura parenquimatosa es normal en esta etapa de la enfermedad incluso con abundante hierro. - Robbins & Kumar Basic Pathology
Páncreas: pigmentación, fibrosis intersticial difusa, atrofia parenquimatosa; hemosiderina en células acinares y de los islotes.
Corazón: cardiomegalia, gránulos de hemosiderina en fibras miocárdicas, coloración parda del miocardio, fibrosis intersticial leve.
Piel: hemosiderina en macrófagos y fibroblastos dérmicos + aumento de melanina epidérmica.
Articulaciones: depósito de hemosiderina en sinovial → sinovitis aguda; depósito de pirofosfato cálcico → pseudogota.
- Robbins & Kumar Basic Pathology, p. 614
Diagnóstico
| Estudio | Hallazgo |
|---|---|
| Saturación de transferrina (ST = hierro sérico/CTFH) | >45% (screening inicial; >35% en mujeres premenopáusicas) |
| Ferritina sérica elevada | Marcador de sobrecarga, guía biopsia y seguimiento |
| Genotipificación de HFE | Confirma diagnóstico si ST >45% + ferritina elevada |
| Biopsia hepática | Reservada para: PFH alteradas, ferritina >1000 μg/L, o edad >40 años |
| Tinción Prussian blue | Hierro hepatocelular visible en azul |
- Andrews' Diseases of the Skin, p. 999
Tratamiento
-
Flebotomía regular es el tratamiento de todos los tipos de hemocromatosis hasta alcanzar niveles de hierro satisfactorios. Con flebotomía periódica y diagnóstico precoz, la esperanza de vida es normal.
-
Evitar vitamina C (puede empeorar la enfermedad).
-
Evitar mariscos crudos (riesgo de infección por Vibrio vulnificus).
-
Cribado familiar de los familiares de primer grado (probandos).
-
Los heterocigotos acumulan hierro en exceso pero generalmente no alcanzan niveles con daño tisular significativo.
-
La flebotomía puede prevenir la cirrosis; sin embargo, una vez establecida la cirrosis, no previene el carcinoma hepatocelular.
-
El daño celular no letal por hierro es reversible y la eliminación del exceso de hierro promueve recuperación funcional.
-
Robbins & Kumar Basic Pathology, p. 615 | Andrews' Diseases of the Skin, p. 1000
Literatura Reciente (2024-2026)
- Una revisión sistemática de 2025 (PMID 38752605) describió en detalle las manifestaciones dermatológicas de la hemocromatosis hereditaria, consistente con lo expuesto arriba.
- Un meta-análisis de 2024 (PMID 39643614) identificó nuevos loci genéticos y consecuencias biomédicas de la variación en la homeostasis del hierro, sugiriendo que la arquitectura genética de la regulación del hierro es más compleja de lo actualmente reconocido.
Fuentes: Robbins & Kumar Basic Pathology (10.ª ed.), p. 613-615 | Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 787-789 | Andrews' Diseases of the Skin, p. 999-1000 | Fitzpatrick's Dermatology | Henry's Clinical Diagnosis by Laboratory Methods
This is a shared conversation. Sign in to Orris to start your own chat.