Pathogenesis of Septic Shock
"septic shock"[MeSH Terms] AND "pathogenesis"[tiab]
Pathogenesis of Septic Shock
Overview
Pathogenic Mechanism Diagram
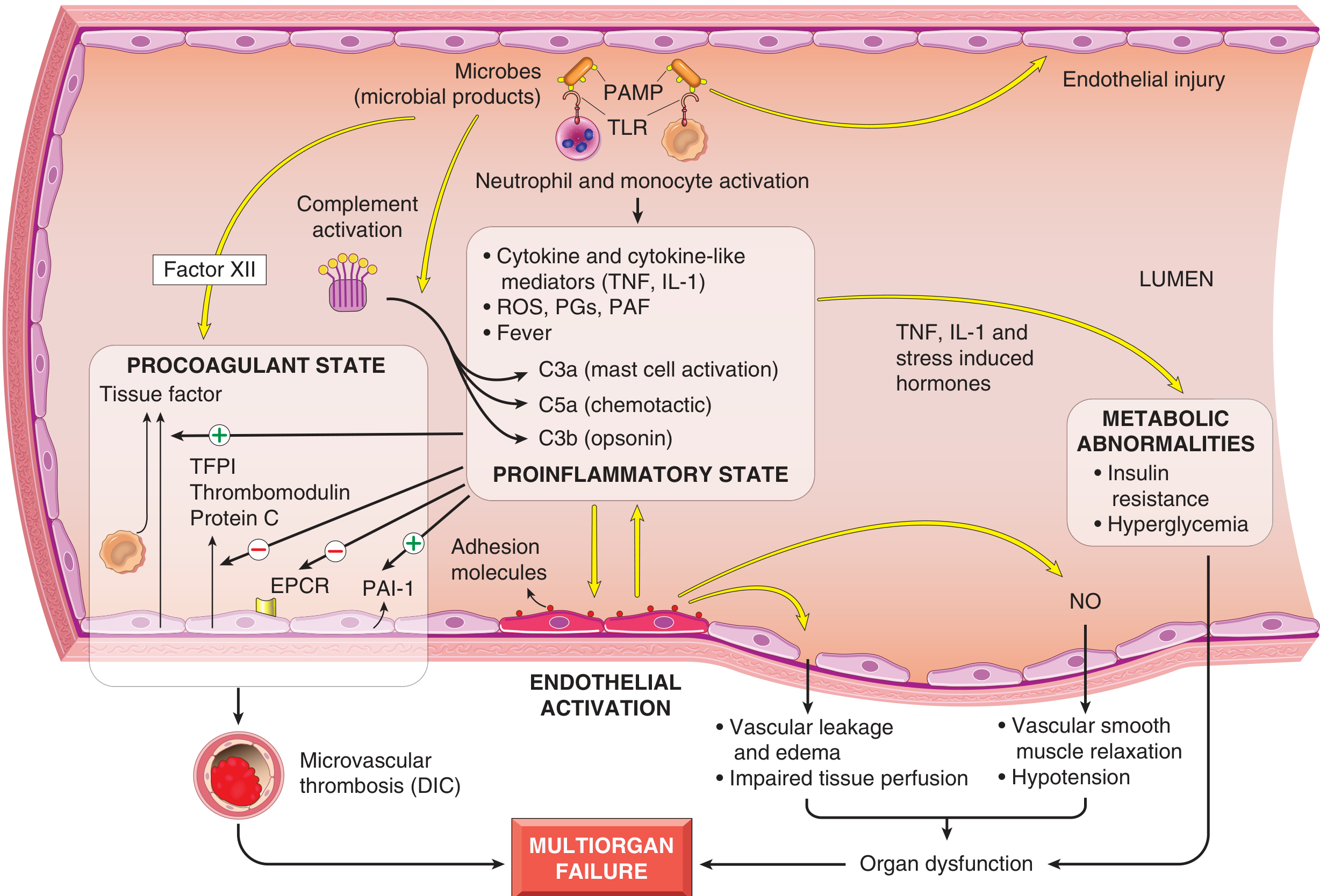
Four Major Pathogenic Pillars
1. Inflammatory and Counter-inflammatory Responses
- Toll-like receptors (TLRs) - detect pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs)
- G-protein-coupled receptors - detect bacterial peptides
- C-type lectin receptors (e.g., Dectins) - detect fungal cell wall components
- Cytokines: TNF, IL-1, IL-12, IL-18, IFN-γ
- High-mobility group box 1 protein (HMGB1)
- Reactive oxygen species (ROS)
- Lipid mediators: prostaglandins, platelet-activating factor (PAF)
- Acute phase reactants: CRP, procalcitonin
- C3a, C5a (anaphylatoxins) - mast cell activation, vasodilation
- C5a (chemotactic fragment) - neutrophil recruitment
- C3b (opsonin) - phagocytosis
- Cytokine shift from Th1 (pro-inflammatory) to Th2 (anti-inflammatory)
- Production of anti-inflammatory mediators: soluble TNF receptor, IL-1 receptor antagonist, IL-10
- Lymphocyte apoptosis and cellular anergy
- Immunosuppressive effects of apoptotic cells
The intensity of pro- vs. anti-inflammatory responses depends on host genetics, underlying disease, pathogen virulence, and burden - Robbins Pathologic Basis of Disease, p. 136
2. Endothelial Activation and Injury
- Pro-inflammatory cytokines loosen endothelial tight junctions → protein-rich edema accumulates throughout the body → impaired tissue perfusion
- Activated endothelium upregulates adhesion molecules → increased leukocyte trafficking
- Upregulation of nitric oxide (NO) production → vascular smooth muscle relaxation → systemic hypotension
- C3a, C5a, and PAF contribute further to vasodilation and hypotension
- Increased capillaries with intermittent flow
- Heterogeneity of flow across capillary beds
- Loss of normal autoregulation of flow based on tissue metabolic environment
- Result: mismatch between oxygen delivery and oxygen needs
3. Induction of a Procoagulant State (DIC)
| Factor | Effect |
|---|---|
| Pro-inflammatory cytokines | Increase tissue factor production by monocytes and endothelial cells |
| Cytokines | Decrease endothelial anticoagulants: TFPI, thrombomodulin, Protein C |
| PAI-1 upregulation | Dampens fibrinolysis |
| Vascular leak + edema | Reduces blood flow in small vessels → stasis → accumulation of activated coagulation factors |
| NETs (neutrophil extracellular traps) | Stimulate both intrinsic and extrinsic coagulation pathways |
4. Metabolic Abnormalities
- Insulin resistance and hyperglycemia: Driven by TNF, IL-1, stress hormones (glucagon, growth hormone, glucocorticoids), and catecholamines stimulating gluconeogenesis, while simultaneously suppressing insulin release and promoting hepatic insulin resistance
- Mitochondrial dysfunction: Oxidative stress causes mitochondrial damage, impairing oxygen utilization even when delivery is maintained
5. Organ Dysfunction and Multiorgan Failure
- Systemic hypotension + interstitial edema + microvascular dysfunction + small vessel thrombosis → decreased O2 and nutrient delivery
- Mitochondrial damage → cells fail to use delivered oxygen
- High cytokine levels → diminished myocardial contractility and reduced cardiac output
- Increased vascular permeability + endothelial injury → Acute Respiratory Distress Syndrome (ARDS)
Superantigens - A Special Case
Stages of Shock (Progressive)
| Stage | Characteristics |
|---|---|
| Nonprogressive | Compensatory mechanisms active (baroreceptors, catecholamines, RAAS, ADH); tachycardia, peripheral vasoconstriction; vital organ perfusion maintained |
| Progressive | Widespread tissue hypoxia; anaerobic glycolysis → lactic acidosis → arteriolar dilation → blood pooling → further reduced cardiac output; endothelial anoxic injury; DIC risk rises |
| Irreversible | Cellular and tissue injury so severe that survival impossible even if hemodynamics corrected; multiorgan failure |
Why Targeted Therapies Have Failed
- Antibiotics to treat the underlying infection
- IV fluids, vasopressors to maintain blood pressure
- Supplemental oxygen to limit tissue hypoxia
molecular basis of carcinogensis
Molecular Basis of Carcinogenesis
Core Principles
- Environmental exposures (chemicals, radiation, viruses)
- Inherited germline mutations
- Spontaneous/random ("bad luck") events
- Endogenous agents (reactive oxygen species, oncometabolites)
- Growth-promoting proto-oncogenes
- Growth-inhibiting tumor suppressor genes
- Apoptosis-regulating genes
- DNA repair genes
Hallmarks of Cancer
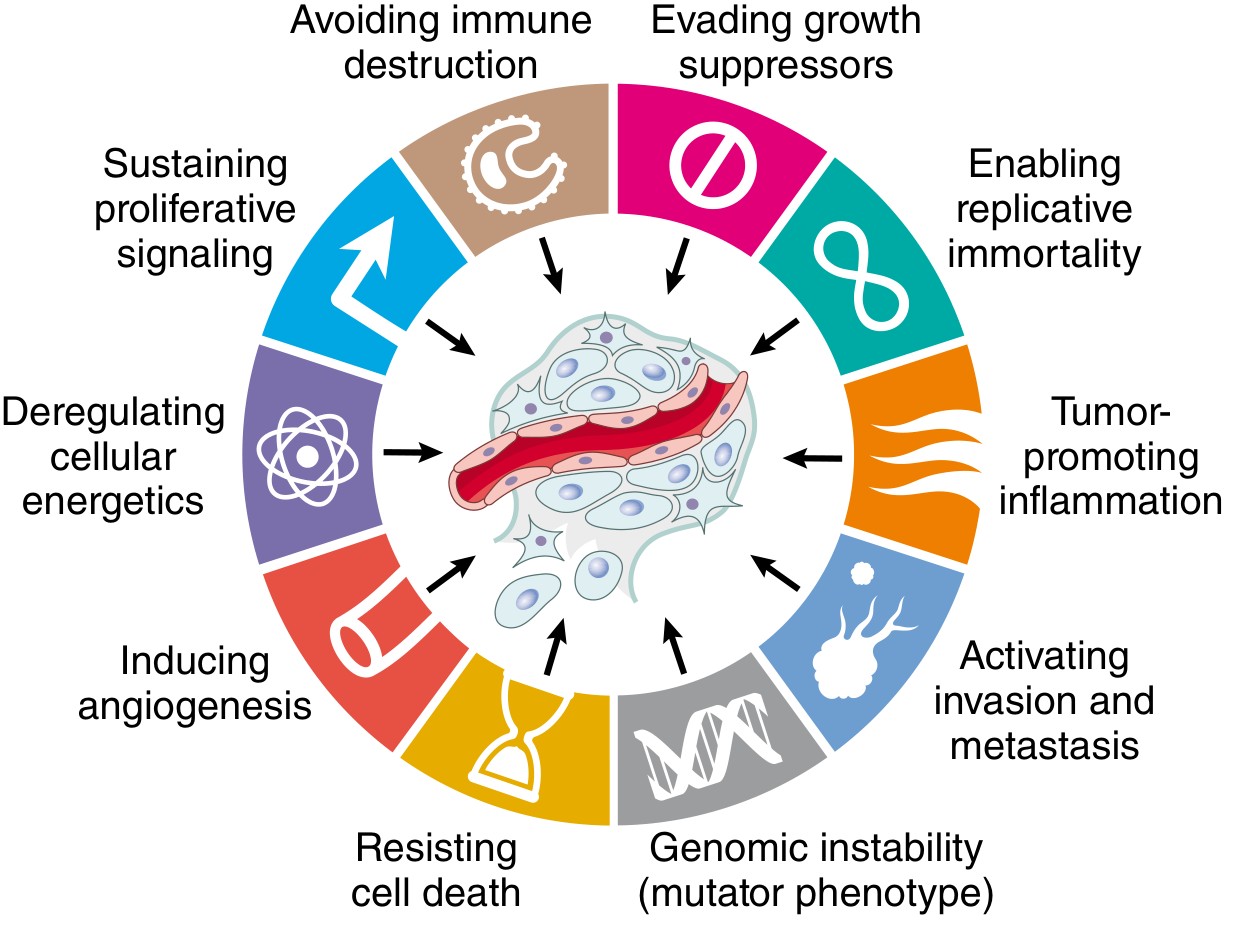
Hallmark 1: Self-Sufficiency in Growth Signals - Oncogenes
Normal Growth Signaling Cascade (co-opted by oncogenes):
- Growth factor binds receptor
- Receptor tyrosine kinase transiently activates
- Cytoplasmic signal transducers relay signal
- Transcription factors are activated in the nucleus
- Growth-promoting genes are expressed → cell division
Key Oncoproteins:
- PDGF-β chain: overexpressed in astrocytomas (autocrine loop)
- FGF: overexpressed in stomach and bladder carcinomas
- ERBB2 (HER2/NEU): amplified in ~25% of breast and ovarian carcinomas; therapeutic target for trastuzumab
- RET: point mutations in MEN2A/2B, papillary thyroid carcinoma
- FLT3: mutated in AML; constitutively activates JAK/STAT signaling
- RAS: most commonly mutated oncogene in human tumors (~30%). Point mutations impair GTPase activity, locking RAS in active GTP-bound state → continuous proliferative signaling. Activated in lung, colon, pancreatic carcinomas (KRAS)
- BRAF (V600E): mutation activates MAPK pathway; found in ~60% of melanomas, papillary thyroid carcinoma; targetable with vemurafenib
- BCR-ABL fusion: translocation t(9;22) - Philadelphia chromosome in CML - constitutively active ABL kinase; targeted by imatinib
- JAK2 V617F: myeloid neoplasms (polycythemia vera); relieves cells of erythropoietin dependence; targeted by ruxolitinib
- All signal transduction pathways converge on nuclear transcription factors
- MYC: most commonly dysregulated transcription factor in cancer; promotes:
- D cyclin expression → cell cycle progression
- Ribosomal RNA synthesis → protein synthesis capacity
- Metabolic reprogramming (Warburg effect)
- Telomerase expression → immortality
- Stem cell reprogramming
- Burkitt lymphoma: virtually always has MYC translocation t(8;14)
Mechanisms of Proto-oncogene Activation:
| Mechanism | Example |
|---|---|
| Point mutation | RAS in colon/lung/pancreatic cancer |
| Gene amplification | HER2 in breast cancer; N-MYC in neuroblastoma |
| Chromosomal translocation (promoter substitution) | MYC in Burkitt lymphoma (t[8;14]) |
| Chromosomal translocation (fusion protein) | BCR-ABL in CML (Philadelphia chromosome t[9;22]) |
Hallmark 2: Insensitivity to Growth Inhibition - Tumor Suppressor Genes
Exception: Haploinsufficiency - in some genes, loss of just one allele is sufficient because normal function requires two doses.
RB: Master Regulator of the Cell Cycle
- In quiescent cells: RB is hypophosphorylated and binds/inhibits E2F transcription factors, blocking S-phase entry
- With growth signals: cyclin D-CDK4/6 phosphorylates RB → releases E2F → cell enters S phase
- Cancer disrupts this via: RB mutation (retinoblastoma, osteosarcoma), CDK4 amplification, cyclin D overexpression, p16INK4a loss
- Viral oncoproteins (HPV E7, adenovirus E1A, SV40 large T antigen) bind and inactivate RB
- RB is functionally disabled in virtually all human cancers through one mechanism or another
TP53: Guardian of the Genome
- DNA damage
- Oncogene activation
- Hypoxia
- Nucleotide depletion
- Normally p53 is kept low by MDM2 (E3 ubiquitin ligase that promotes p53 degradation)
- Stress signals (ATM/ATR kinases) phosphorylate p53 → releases from MDM2 → p53 stabilizes
- Active p53 upregulates:
- p21 (CDK inhibitor) → G1/S arrest → time for DNA repair
- GADD45 → DNA repair
- BAX → apoptosis
- Senescence programs if damage is irreparable
- Inactivated by HPV E6 protein (accelerates p53 degradation)
APC: Gatekeeper of Colonic Neoplasia
- APC protein forms a "destruction complex" (with Axin and GSK-3β) that phosphorylates β-catenin for proteasomal degradation
- Loss of APC → β-catenin accumulates → translocates to nucleus → forms TCF complex → activates MYC, cyclin D1, and other progrowth genes
- Tumors with normal APC often instead have activating β-catenin mutations
Other Key Tumor Suppressors:
| Gene | Protein | Function | Familial Syndrome |
|---|---|---|---|
| PTEN | PTEN phosphatase | Inhibits PI3K/AKT signaling | Cowden syndrome |
| VHL | VHL protein | Inhibits HIF-1α (hypoxia-induced transcription) | VHL syndrome, renal cell carcinoma |
| CDKN2A | p16INK4a / p14ARF | CDK inhibitor / p53 stabilizer | Familial melanoma |
| SMAD2/4 | SMAD proteins | TGF-β signaling (growth inhibitory) | Colorectal, pancreatic carcinoma |
| NF1 | Neurofibromin-1 | Inhibitor of RAS/MAPK signaling | Neurofibromatosis type 1 |
| BRCA1/2 | BRCA proteins | DNA repair (homologous recombination) | Familial breast/ovarian cancer |
Hallmark 3: Evasion of Apoptosis
- BCL2 overexpression: t(14;18) translocation in follicular lymphoma places BCL2 under Ig heavy chain promoter → BCL2 overexpressed → blocks cytochrome c release from mitochondria → apoptosis blocked
- Loss of BAX (pro-apoptotic)
- Loss of p53 (major inducer of apoptosis)
- Anoikis resistance: tumor cells resist apoptosis triggered by loss of matrix attachment (mediated by altered integrin expression)
Hallmark 4: Limitless Replicative Potential (Immortality)
- Normal cells divide 60-70 times then permanently exit the cell cycle (senescence)
- Senescence driven by p53 and p16/INK4a maintaining RB hypophosphorylated
- Cancer cells bypass senescence via RB/p53 pathway disruption
- Cells that bypass senescence still die via progressive telomere shortening
- When telomeres are eroded: exposed chromosome ends trigger DNA damage response → if p53 is intact, apoptosis; if p53 is lost, breakage-fusion-bridge cycles cause catastrophic genomic damage
- Cancer cells that survive crisis must reactivate telomerase
- 85-95% of tumors express telomerase; remainder use alternative lengthening of telomeres (ALT) via DNA recombination
- Tissue stem cells naturally express telomerase and have self-renewal capacity
- Cancer may arise from stem cells or from differentiated cells that acquire stem-like properties
- Cancer stem cells are the source of tumor recurrence and therapy resistance
Hallmark 5: Altered Cellular Metabolism - Warburg Effect
- Oxidative phosphorylation converts glucose entirely to CO₂ + H₂O → no carbon available for biosynthesis
- Aerobic glycolysis provides carbon intermediates for synthesis of DNA, proteins, lipids, and organelles needed for cell division
- Glutamine also provides carbon via the TCA cycle for lipid biosynthesis (citrate → acetyl-CoA)
Hallmark 6: Sustained Angiogenesis
- VEGF (Vascular Endothelial Growth Factor) is the dominant pro-angiogenic factor
- Upregulated by HIF-1α in hypoxia, by RAS/MYC oncogenes, and released from ECM by MMPs
- VHL tumor suppressor normally targets HIF-1α for degradation - loss of VHL (renal cell carcinoma) → constitutive HIF-1α → VEGF overproduction
- Anti-VEGF therapy (bevacizumab) is approved for multiple cancers
Hallmark 7: Invasion and Metastasis
- Reduced cell-cell adhesion - loss of E-cadherin (tumor suppressor function); gain of N-cadherin (mesenchymal marker) = Epithelial-Mesenchymal Transition (EMT)
- ECM degradation - upregulation of matrix metalloproteinases (MMPs), especially MMP2 and MMP9, which cleave collagen IV and laminin in basement membranes; MMP cleavage products also release VEGF and create new integrin-binding sites
- Altered integrin expression - cancer cells change integrin repertoire to facilitate migration along degraded ECM; resistance to anoikis (loss of normal matrix survival signals)
- Locomotion - driven by autocrine motility factors (chemokines, IGF), matrix cleavage products, stromal cell paracrine factors (HGF/scatter factor acting via MET receptor)
- Intravasation → systemic circulation → extravasation → colonization at distant site (organ tropism governed by chemokine receptor-ligand pairs and pre-metastatic niche)
Hallmark 8: Evasion of Host Immune Response
- Loss of MHC-I expression → invisible to CD8+ cytotoxic T cells
- Loss of tumor antigens → no target for immune recognition
- PD-L1/PD-L2 upregulation → engage PD-1 on T cells → T cell exhaustion/inhibition
- CTLA-4 promotion → neutralizes B7 on APCs → reduces T cell activation
- Immunosuppressive cytokines: TGF-β, IL-10, prostaglandin E2, VEGF (blocks T cell trafficking into tumor)
- Regulatory T cell (Treg) induction → active immune suppression in tumor microenvironment
- Myeloid-derived suppressor cells (MDSCs) in tumor microenvironment
Enabling Characteristic: Genomic Instability
| Pathway | Syndrome | Cancer Risk |
|---|---|---|
| Mismatch repair (MMR) | HNPCC/Lynch syndrome | Colorectal carcinoma; microsatellite instability (MSI) |
| Nucleotide excision repair (NER) | Xeroderma pigmentosum | UV-induced skin cancers; can't repair pyrimidine dimers |
| Homologous recombination | BRCA1/2 mutations, Bloom syndrome, Ataxia-telangiectasia, Fanconi anemia | Breast, ovarian, and other carcinomas |
| DNA polymerase proofreading | POLE/POLD1 mutations | Colorectal and endometrial carcinoma |
- Translocations activate oncogenes by:
- Promoter/enhancer substitution (e.g., MYC brought under Ig promoter in Burkitt lymphoma)
- Fusion proteins with novel activity (e.g., BCR-ABL in CML)
- Amplifications create extra copies of oncogenes (shown as double minutes or homogeneously staining regions on karyotype)
- Deletions remove tumor suppressor genes
- Whole chromosome gains/losses (aneuploidy)
Enabling Characteristic: Epigenetic Alterations
- DNA hypermethylation of CpG islands in promoters of tumor suppressor genes silences them without mutation
- Global DNA hypomethylation activates proto-oncogenes and promotes genomic instability
- Histone modifications (methylation, acetylation) alter chromatin accessibility
- MicroRNAs - can act as oncogenes (oncomirs) by silencing tumor suppressors, or as tumor suppressors by silencing oncogenes
Multistep Carcinogenesis: Putting It All Together
- APC loss (or β-catenin mutation) - initiating event; activates Wnt/MYC/cyclin D1
- KRAS mutation - drives autonomous proliferation
- SMAD4 loss - disables TGF-β growth inhibition
- TP53 loss - eliminates apoptosis/senescence checkpoint
- Additional driver mutations → invasion and metastasis
Ihc of wound healing FMT pg answer for 10 marks
Wound Healing (Forensic Medicine & Toxicology) - 10 Marks
Definition
- Determine whether a wound is ante-mortem, peri-mortem, or post-mortem
- Estimate the age (time) of the wound from time of infliction
- Establish the vital reaction - proof that the individual was alive when the injury was sustained
Types of Wound Healing
1. Healing by First Intention (Primary Union)
- Clean, well-apposed wound margins (e.g., surgical incision)
- Minimal tissue loss, limited inflammation
- Heals with a neat linear scar
- Faster, fewer complications
2. Healing by Second Intention (Secondary Union)
- Large tissue defect, irregular/infected wound
- Marked inflammatory reaction
- Abundant granulation tissue formation
- Wound contraction is prominent (myofibroblasts)
- Heals with contracted, irregular scar; prone to complications
Phases of Wound Healing (with Histological Timelines)
Phase 1: Haemostasis (Immediate - Minutes)
- Vascular injury → vasoconstriction (transient)
- Platelet aggregation → platelet plug formation
- Coagulation cascade activation → fibrin clot
- Fibrin clot acts as provisional scaffold
Phase 2: Inflammatory Phase (Hours to 5 Days)
- Vasodilation and increased vascular permeability
- Neutrophil (PMN) infiltration begins at wound margins within 24 hours - the earliest reliable sign of vital reaction
- Neutrophils phagocytose bacteria and debris
- Thin layer of epithelial cells begins migration
- Monocytes arrive and differentiate into macrophages (appear at 24-48 hours, peak at 48-72 hours)
- Macrophages: orchestrate repair by producing cytokines (TNF, IL-1, TGF-β, PDGF, FGF, VEGF)
- Continued neutrophil infiltration
- Epithelial cells proliferate and cover wound surface
Phase 3: Proliferative Phase / Granulation Tissue Formation (3 Days to 3 Weeks)
- Granulation tissue begins to appear - hallmark feature
- Proliferating fibroblasts migrating into wound
- Neovascularization (angiogenesis) - new thin-walled capillaries
- Loose extracellular matrix (ECM)
- Interspersed macrophages
- Gross: pink, soft, granular appearance
- Histology: fibroblasts + capillaries + inflammatory cells in loose ECM
- Granulation tissue fills the defect
- Fibroblasts synthesize collagen (initially Type III, later Type I)
- Macrophages drive fibroblast activity via TGF-β (most potent fibrogenic cytokine)
- Myofibroblasts appear → wound contraction
- Epithelialization complete
- Progressive reduction in vascularity and cellularity
- Collagen deposition increases
Phase 4: Remodelling Phase (3 Weeks to 1 Year+)
- Granulation tissue replaced by dense fibrous scar
- Type III collagen replaced by Type I collagen (stronger, organized)
- Matrix metalloproteinases (MMPs) remodel ECM, balanced by TIMPs
- Progressive vascular regression → pale, avascular scar
- Tensile strength increases: reaches 70-80% of original strength by 3 months (never fully recovers to 100%)
Kinetics of Wound Healing - Summary Table (for Forensic Age Estimation)
| Time | Histological Events |
|---|---|
| Immediate (0-30 min) | Blood clot formation; platelet plug; fibrin deposition |
| 1-6 hours | Vascular changes; early oedema; no cellular infiltrate yet |
| 6-12 hours | Margination of neutrophils; early PMN infiltration begins |
| 24 hours | Neutrophils at incision margins; epithelial cells begin migration |
| 24-48 hours | Macrophages appear; continuous neutrophil infiltration |
| 48-72 hours | Peak macrophage infiltration; fibroblast proliferation begins |
| 3-5 days | Granulation tissue well established; neovascularization; fibroblasts prominent |
| 5-10 days | Collagen deposition begins; bridging of incision by collagen fibers |
| 2 weeks | Continued collagen accumulation; leukocytes disappear; vascular regression begins |
| 1 month | Dense collagenous scar; no inflammatory cells; sparse vessels |
Vital Reaction - Forensic Significance
| Feature | Ante-mortem | Post-mortem |
|---|---|---|
| Haemorrhage into tissues | Present (soaks tissue, clots) | Absent or minimal (ooze only) |
| Retraction of wound edges | Present | Absent |
| Neutrophil infiltration | Appears >6 hours | Absent |
| Granulation tissue | Appears by day 3-5 | Absent |
| Histamine release | Present | Absent |
| Vital bruising | Present (blue-red, firm) | Post-mortem lividity (purple, skin surface only) |
Factors Affecting Wound Healing
- Infection (most important - prolongs inflammation, delays healing)
- Blood supply / ischaemia
- Foreign bodies (perpetuate chronic inflammation)
- Size and type of wound
- Mechanical factors (mobility, tension)
- Diabetes mellitus (vascular disease, neuropathy, impaired immunity) - most important systemic cause
- Nutritional deficiency - protein and Vitamin C deficiency impair collagen synthesis
- Glucocorticoids/steroids - anti-inflammatory; inhibit TGF-β → weak scar
- Aging (reduced regenerative capacity)
- Anaemia and hypoxia
Abnormal Wound Healing
| Complication | Mechanism | Features |
|---|---|---|
| Keloid | Excess collagen beyond wound margins | Extends beyond original wound; more common in dark skin; earlobes, deltoid, presternal; recurs after excision |
| Hypertrophic scar | Excess collagen within wound margins | Raised, firm; does not extend beyond borders; regresses with time |
| Chronic wound / Ulcer | Failed healing (ischaemia, infection, diabetes) | Continued inflammation; non-healing granulation tissue |
| Wound dehiscence | Inadequate scar formation | Wound pulls apart; seen in infection, nutritional deficiency |
| Contracture | Excess wound contraction by myofibroblasts | Impairs function over joints; common in burns |
Growth Factors in Wound Healing (Key Points)
| Growth Factor | Source | Role |
|---|---|---|
| TGF-β | Macrophages, fibroblasts, platelets | Most potent fibrogenic agent; collagen synthesis; fibroblast proliferation |
| PDGF | Platelets, macrophages | Fibroblast and smooth muscle proliferation; chemotaxis |
| VEGF | Macrophages, fibroblasts | Angiogenesis; vascular permeability |
| FGF | Macrophages, mast cells | Angiogenesis; fibroblast proliferation |
| EGF / TGF-α | Keratinocytes, macrophages | Epithelial proliferation and migration |
Forensic Points to Remember
- Neutrophil infiltration is the earliest reliable microscopic sign of a vital wound reaction (appears ~6-12 hours)
- Granulation tissue by day 3-5 confirms survival of at least 3 days after injury
- A wound showing only fibrin clot with no cellular reaction suggests survival < 6 hours or a peri-mortem wound
- Complete re-epithelialization by 5-7 days (first intention) can estimate post-operative survival time in medicolegal autopsies
- Post-mortem wounds show no vital reaction - no tissue retraction, no haemorrhage into tissues, no inflammatory infiltrate
Create a summary table comparing wound age estimation markers for FMT exams
Wound Age Estimation Markers - Summary Table for FMT Exams
Master Table: Histological Markers vs. Time Since Infliction
| Time Since Injury | Phase | Dominant Cells | Vascular Changes | Connective Tissue Changes | Epithelial Changes | Forensic Inference |
|---|---|---|---|---|---|---|
| 0-30 min | Haemostasis | Platelets | Vasoconstriction → vasodilation; transudation | Fibrin clot formed; platelet plug | None | Wound inflicted ante-mortem (vital); survival < 1 hour |
| 1-6 hours | Early inflammation | RBCs leaking; early PMN margination | Increased permeability; oedema | Fibrin network forming | None | Wound is ante-mortem; very recent |
| 6-12 hours | Early inflammation | Neutrophils begin infiltrating wound margins | Vascular congestion; oedema | Fibrin clot intact | None | Earliest reliable cellular vital reaction; survival ~6-12 hours |
| 24 hours | Acute inflammation | Neutrophils prominent at wound margins | Active hyperaemia; oedema | Fibrin clot; no collagen yet | Basal keratinocytes begin migrating; mitotic activity increased | Confirms survival of at least 24 hours |
| 24-48 hours | Acute inflammation | Neutrophils peak; monocytes arrive | Continued hyperaemia | Fibrin still dominant scaffold | Epithelial cells from both edges proliferate - thin continuous layer forming | Survival 24-48 hours |
| Day 2-3 | Transition to repair | Macrophages replace neutrophils (M1-like: IL-1β, TNF-α, IL-12) | Early angiogenesis initiated | ECM deposition begins; loose stroma | Epidermal migration continues | Transition to proliferative phase; survival ≥ 2-3 days |
| Day 3-5 | Proliferative | Macrophages dominant; fibroblasts appear | Granulation tissue forming - new thin-walled capillaries (angiogenesis) | Loose collagen; fibroblasts proliferating; granulation tissue well established | Re-epithelialization progressing | Granulation tissue = survival ≥ 3 days; key FMT marker |
| Day 5-7 | Proliferative | Fibroblasts prominent; macrophages (M2 anti-inflammatory: IL-4, IL-10, TGF-β) | Maximum neovascularization; abundant granulation tissue | Collagen Type III deposited; ECM deposition active | Epidermal layer restored (1st intention wounds) | Survival ≥ 5-7 days; wound well into repair |
| Day 7-14 | Proliferative / early remodelling | Fibroblasts; myofibroblasts appear (wound contraction) | Vascularity begins regressing | Continued collagen deposition; fibroblast proliferation | Epithelialization complete | Myofibroblasts indicate survival ≥ 1 week |
| Day 14-30 | Remodelling | Fibroblasts decreasing; few inflammatory cells | Progressive vascular regression; vessels disappear | Dense collagen accumulation; maturing scar; Type III → Type I collagen | Epidermis normal | Dense collagen without inflammation = wound > 2 weeks old |
| Day 30+ | Scar maturation | Sparse fibroblasts; no inflammatory cells | Few or no vessels (avascular scar) | Dense, organized Type I collagen bundles; acellular; pale scar | Normal epidermis | Complete scar = survival ≥ 1 month |
| Months - 1 year | Final remodelling | Virtually acellular | Avascular | Collagen cross-linking; tensile strength 70-80% of normal | Normal | Old healed scar |
Quick-Reference Card: Key Forensic Milestones
| Event | Earliest Appearance | Peak | Gone By |
|---|---|---|---|
| Platelet plug / fibrin clot | Immediate (minutes) | 1-6 hours | Day 3-5 (dissolved) |
| Oedema | 1-2 hours | 24-48 hours | Day 5 |
| Neutrophil (PMN) infiltration | ~6-12 hours | 24-48 hours | Day 3 (wane) |
| Macrophage infiltration | 24-48 hours | Day 3-5 | Day 10-14 |
| Early granulation tissue | Day 3 | Day 5-7 | Day 21-30 |
| Fibroblast proliferation | Day 2-3 | Day 7-14 | Day 21+ |
| Myofibroblasts (wound contraction) | Day 5-7 | Day 7-14 | Day 21-28 |
| New capillaries (angiogenesis) | Day 3 | Day 5-7 | Day 14-21 |
| Collagen Type III | Day 3-5 | Day 7-14 | Gradually replaced |
| Collagen Type I (mature) | Day 7-10 | Day 21-30 | Persists (permanent scar) |
| Re-epithelialization complete | Day 5-7 (1st intention) | - | - |
| Dense acellular scar | Day 30 | Month 3 | Never (permanent) |
Vital Reaction Markers: Ante-mortem vs. Post-mortem Wounds
| Feature | Ante-mortem Wound | Post-mortem Wound | Forensic Significance |
|---|---|---|---|
| Haemorrhage into tissues | Present; blood soaks tissue; clots | Absent or ooze only (no clot) | Vital haemorrhage confirms ante-mortem |
| Retraction of wound edges | Present (elastic tissue recoil) | Absent | Retraction = vital reaction |
| Neutrophil infiltration | Present (>6-12 hours post-infliction) | Absent | Most important early cellular marker |
| Macrophages | Present (>24-48 hours) | Absent | Confirms survival after injury |
| Granulation tissue | Present (>3 days) | Absent | Confirms survival ≥ 3 days |
| Oedema | Present (inflammatory exudate) | Absent | Supports ante-mortem |
| Histamine release | Present (from mast cells) | Absent | Biochemical vital reaction |
| Vital bruising | Blue-red; firm; in deep tissue | Purplish; skin surface only (lividity) | Key external examination point |
| RBC extravasation | Present with leucocyte infiltration | RBCs only; no leucocytes | Leucocytes confirm vital reaction |
| Fibrin | Present in vessel walls and tissues | Absent | Vital reaction |
Phases at a Glance: One-Line Summary
| Phase | Timing | Hallmark Finding | Key Mediator |
|---|---|---|---|
| Haemostasis | 0 min - 6 hours | Platelet plug + fibrin clot | Thromboxane A2, thrombin |
| Inflammation | 6 hours - 5 days | Neutrophils → Macrophages | TNF-α, IL-1, IL-6 |
| Proliferation | Day 3 - 3 weeks | Granulation tissue; angiogenesis; fibroblasts | TGF-β, VEGF, PDGF, FGF |
| Remodelling | 3 weeks - 1 year | Type III → Type I collagen; scar maturation | MMPs, TIMPs, TGF-β |
Factors That Alter Wound Age Estimation (FMT Caveat Points)
| Factor | Effect on Healing Rate | FMT Implication |
|---|---|---|
| Infection | Delays (prolongs inflammation) | Neutrophils persist longer; misleading age estimate |
| Diabetes mellitus | Delays (vascular + immune impairment) | Granulation tissue appears late |
| Steroids/glucocorticoids | Delays (suppresses inflammation + TGF-β) | Few inflammatory cells; misleading early age |
| Malnutrition / Vit C deficiency | Delays (impairs collagen synthesis) | Weak or absent collagen deposition |
| Ageing | Delays | All phases slower |
| Good blood supply / clean wound | Accelerates | Normal or faster timeline |
| Antibiotics | Modifies inflammatory phase | Infection-related delay prevented |
Memory Aid: "NEGA-FICS"
| Letter | Marker | Timing |
|---|---|---|
| N | Neutrophils | 6-24 hours |
| E | Epithelial migration | 24-48 hours |
| G | Granulation tissue | Day 3-5 |
| A | Angiogenesis (peak) | Day 5-7 |
| F | Fibroblasts (peak) | Day 7-14 |
| I | collagen type III | Day 3-14 |
| C | Collagen type I (mature) | Day 10-30+ |
| S | Scar (complete, acellular) | Day 30+ |
Circle of Willis and it’s Medicolegal importance
Circle of Willis and Its Medicolegal Importance
Definition
Anatomy
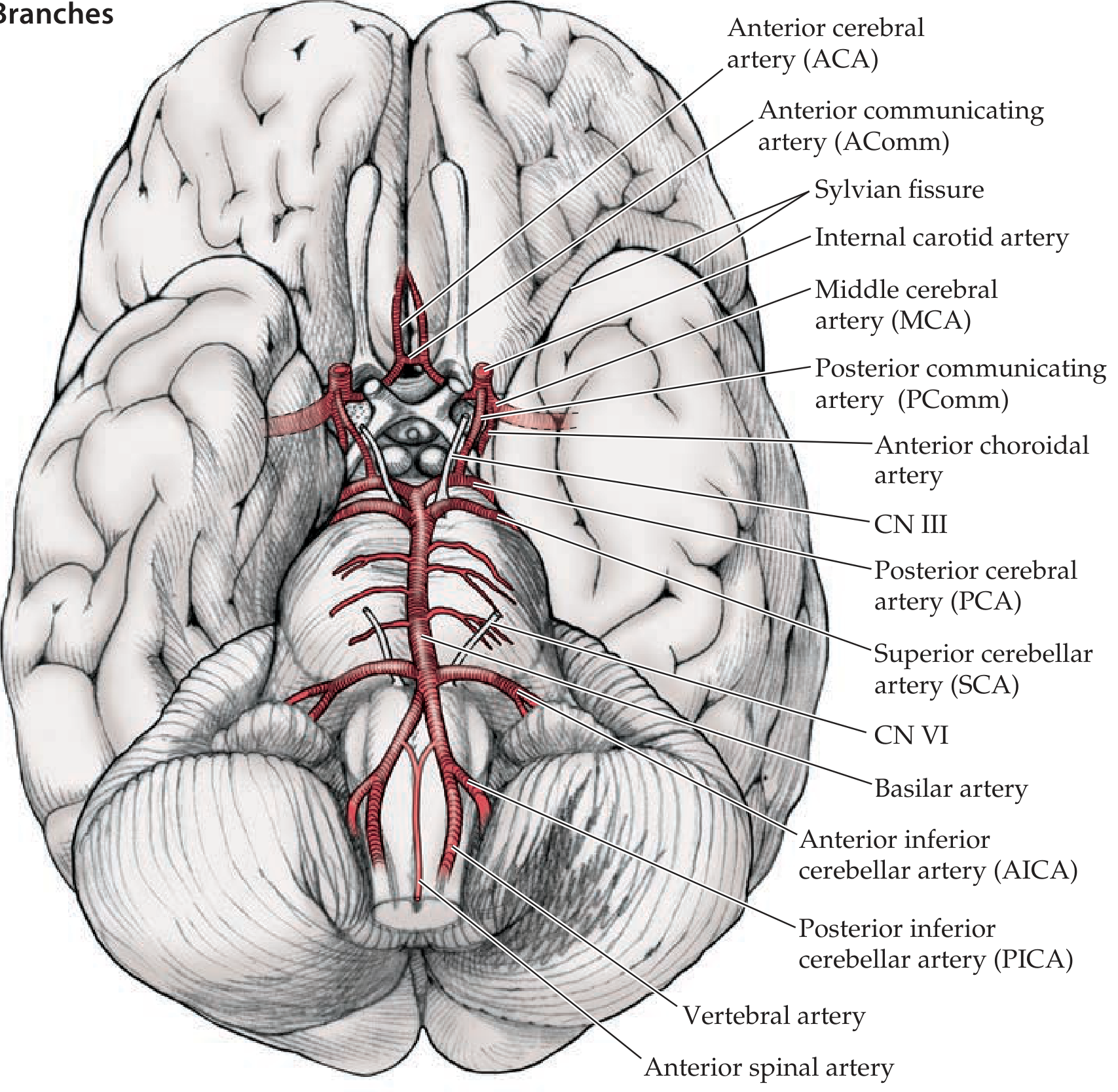
Vessels Forming the Circle (Anterior → Posterior)
- Anterior Cerebral Arteries (ACA) - bilateral; terminal branches of ICAs
- Anterior Communicating Artery (AComm) - single; connects the two ACAs anteriorly
- Internal Carotid Arteries (ICA) - bilateral; enter the circle after giving off ophthalmic, anterior choroidal, and PComm branches
- Middle Cerebral Arteries (MCA) - bilateral; largest terminal branch of ICA (exits the circle laterally, not strictly part of the ring)
- Posterior Communicating Arteries (PComm) - bilateral; connect the ICA to the PCA, linking anterior and posterior circulations
- Vertebral arteries - arise from subclavian arteries; ascend through foramina transversaria of cervical vertebrae; enter foramen magnum
- Basilar artery - formed by fusion of the two vertebral arteries at the pontomedullary junction
- Posterior Cerebral Arteries (PCA) - bilateral; terminal branches of the basilar artery; complete the circle posteriorly
Summary Table: Components
| Vessel | Number | Origin | Connects |
|---|---|---|---|
| Anterior Cerebral Artery (ACA) | 2 (bilateral) | ICA | Anterolateral part of ring |
| Anterior Communicating Artery (AComm) | 1 | Connects both ACAs | Anterior midline of ring |
| Internal Carotid Artery (ICA) | 2 (bilateral) | Common carotid | Forms lateral limbs |
| Posterior Communicating Artery (PComm) | 2 (bilateral) | ICA → PCA | Links anterior and posterior circulations |
| Posterior Cerebral Artery (PCA) | 2 (bilateral) | Basilar artery | Posterior part of ring |
| Basilar artery | 1 | Vertebral arteries | Midline posterior |
Functional Significance
- Collateral blood flow: If one feeder artery is occluded, blood can be rerouted via the circle to maintain perfusion of the affected hemisphere
- Equalises pressure between the two sides of the cerebral circulation
- Complete ring in only ~34% of individuals - anatomical variants (hypoplastic or absent communicating arteries) are common, reducing the effectiveness of collateral flow
- Supplies all three major cerebral arteries from which the entire cerebral cortex is perfused:
- ACA → medial frontal and parietal lobes
- MCA → lateral cerebral hemisphere (largest territory)
- PCA → occipital lobes and inferior temporal lobes
Medicolegal Importance
1. Berry (Saccular) Aneurysm and Subarachnoid Haemorrhage (SAH)
- Thin-walled saccular outpouching, almost always at arterial branch points in or just beyond the circle
- Wall lacks smooth muscle and internal elastic lamina (congenital structural defect in the tunica media)
- Endothelial dysfunction from haemodynamic stress at bifurcation points drives progressive dilatation
- Found in about 2% of the population; multiple aneurysms in 20-30% of cases
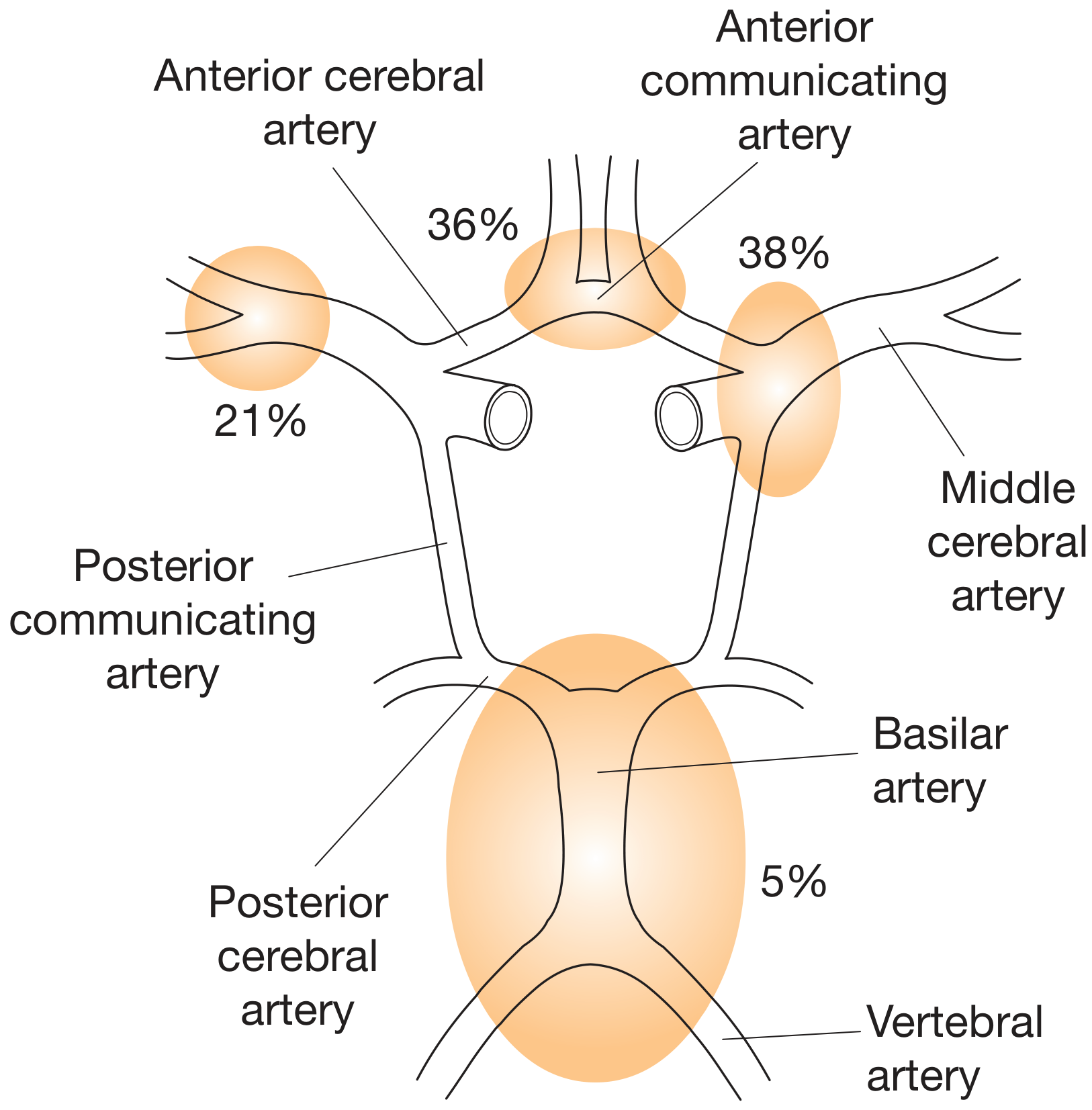
| Site | Frequency |
|---|---|
| Anterior communicating artery (AComm) | 38% (most common) |
| Anterior cerebral artery (ACA) | 36% |
| Middle cerebral artery (MCA) | 21% |
| Vertebrobasilar junction | 5% |
- Thin-walled, bright red, shiny outpouching (few mm to 2-3 cm)
- Wall: thickened hyalinized intima + adventitia only (no media or elastic lamina)
- Rupture occurs at the apex of the sac
- Blood extravasates into subarachnoid space and/or brain parenchyma
- Classic "thunderclap" headache - sudden, excruciating ("worst headache of my life")
- Loss of consciousness (50%)
- Vomiting (70%)
- Seizure (10%)
- Neck stiffness and photophobia (meningism) - develop over hours
- Painful 3rd nerve palsy (CN III compression) - characteristic of posterior communicating artery (PComm) aneurysm
- Subhyaloid haemorrhages on fundoscopy (Terson syndrome)
- Rupture rate: 1.3% per year overall; aneurysms >10 mm have ~50% annual rupture risk
- 25-50% of patients die with the first rupture - sudden unexpected death
- One-third of cases misdiagnosed initially as tension/migraine headache - medicolegal liability
- Commonly occurs with acute intracranial pressure rises (straining at stool, sexual exertion, heavy lifting) - important for establishing circumstances of death
- Predisposing conditions with medicolegal significance:
- Autosomal dominant polycystic kidney disease (ADPKD)
- Ehlers-Danlos syndrome type IV
- Marfan syndrome
- Neurofibromatosis type 1 (NF1)
- Coarctation of the aorta
- Hypertension (in ~50% of cases)
- Smoking, cocaine abuse
2. Subarachnoid Haemorrhage (SAH) - Forensic Autopsy Findings
- Subarachnoid space filled with blood over the base of the brain and in the basal cisterns
- Thick blood clot concentrated around the circle of Willis
- Site of aneurysmal rupture may be identifiable
- Brain swelling with possible herniation
- Vasospasm (days 4-14 post-SAH) - involving vessels of the circle of Willis; mediated by endothelins, NO, and arachidonic acid metabolites → secondary infarction
- Communicating hydrocephalus - meningeal fibrosis and scarring obstruct CSF flow and resorption (late complication)
- Hemosiderin-laden macrophages indicate prior haemorrhage (age of bleed estimation)
3. Medicolegal Importance: Death Certification and COD
| Scenario | Medicolegal Issue |
|---|---|
| Young person found dead after exertion / straining | Consider ruptured berry aneurysm; brain base examination mandatory |
| "Worst headache" misdiagnosed as migraine | Medical negligence; delayed diagnosis |
| Blunt head trauma with SAH | Distinguish traumatic SAH from spontaneous ruptured aneurysm |
| Death during sexual activity or physical effort | COD: ruptured berry aneurysm; mode of death: natural |
| Known polycystic kidney disease | Predisposition to intracranial aneurysm; requires disclosure |
| Sudden death in hypertensive patient | SAH vs. hypertensive intracerebral haemorrhage - needs autopsy |
4. Anatomical Variants and Medicolegal Significance
- Complete, full-caliber Circle of Willis present in only ~34% of individuals
- Hypoplastic or absent PComm - most common variant; reduces posterior-anterior collateral flow
- Asymmetric ICAs or ACAs - important in determining stroke territory
- Fenestrated arteries and duplications at branch points - sites of turbulent flow and aneurysm formation
- Variants affect outcome after surgical clipping or endovascular coiling - surgical complication liability
5. Other Medicolegal Associations
- Mostly affects the basilar artery
- More common in elderly hypertensives
- Causes ischaemic strokes rather than SAH
- May follow blunt or penetrating head trauma
- Important to distinguish from spontaneous aneurysm in alleged assault cases
- Infective infiltration of arterial wall (infective endocarditis, IV drug use)
- May occur in peripheral branches near/beyond the circle
- Important in medicolegal cases involving IV drug abusers
- Occlusion of vessels at or arising from the circle of Willis produces specific territorial infarcts
- ACA territory: contralateral leg weakness
- MCA territory: contralateral hemiplegia + aphasia (dominant side)
- PCA territory: contralateral homonymous hemianopia
- Important for determining disability, causation, and negligence claims
Summary: Key Points for FMT Exams
| Point | Detail |
|---|---|
| Location | Base of brain, interpeduncular cistern |
| Formed by | ICA (bilateral) + Basilar artery via vertebrals |
| Communicating arteries | AComm (1) + PComm (2) |
| Complete ring | Only ~34% of population |
| Aneurysm prevalence | ~2% of general population |
| Commonest aneurysm site | AComm (38%) |
| Presentation of rupture | Thunderclap headache + LOC |
| Mortality with first rupture | 25-50% |
| Vasospasm peak | Day 4-14 post-SAH |
| Forensic importance | Sudden natural death; trauma vs. spontaneous SAH; medical negligence |
| Predisposing diseases | ADPKD, Marfan, Ehlers-Danlos, NF1, coarctation, hypertension |
Generate a summary PDF on berry aneurysm pathology and morphology
ls /home/daytona/workspace/ 2>/dev/null || mkdir -p /home/daytona/workspace
mkdir -p /home/daytona/workspace/berry-aneurysm && curl -s -o /home/daytona/work…
~/berry-aneurysm/generate_pdf.py
from reportlab.lib.pagesizes import A4
from reportlab.lib import colors
from reportlab.lib.units import cm, mm
from reportlab.lib.styles import getSampleStyleSheet, ParagraphStyle
from reportlab.lib.enums import TA_CENTER, TA_LEFT, TA_JUSTIFY
from reportlab.platypus import (
SimpleDocTemplate, Paragraph, Spacer, Table, TableStyle,
HRFlowable, Image, KeepTogether, PageBreak
)
from reportlab.platypus.flowables import HRFlowable
from reportlab.pdfgen import canvas
from reportlab.lib.colors import HexColor
import os
# ── Colour palette ──────────────────────────────────────────────────
DEEP_RED = HexColor("#8B1A1A")
DARK_RED = HexColor("#C0392B")
LIGHT_RED = HexColor("#FADBD8")
PALE_GREY = HexColor("#F4F6F7")
MID_GREY = HexColor("#BDC3C7")
DARK_GREY = HexColor("#2C3E50")
ACCENT_BLUE = HexColor("#2E86AB")
WHITE = colors.white
BLACK = colors.black
OUTPUT = "/home/daytona/workspace/berry-aneurysm/Berry_Aneurysm_Summary.pdf"
# ── Page canvas callbacks (header / footer) ─────────────────────────
def on_page(canvas_obj, doc):
W, H = A4
# Top rule
canvas_obj.setStrokeColor(DEEP_RED)
canvas_obj.setLineWidth(2)
canvas_obj.line(1.5*cm, H - 1.2*cm, W - 1.5*cm, H - 1.2*cm)
# Footer
canvas_obj.setFont("Helvetica", 7)
canvas_obj.setFillColor(MID_GREY)
canvas_obj.drawString(1.5*cm, 0.8*cm, "Berry Aneurysm – Pathology & Morphology Summary")
canvas_obj.drawRightString(W - 1.5*cm, 0.8*cm, f"Page {doc.page}")
canvas_obj.setStrokeColor(MID_GREY)
canvas_obj.setLineWidth(0.5)
canvas_obj.line(1.5*cm, 1.2*cm, W - 1.5*cm, 1.2*cm)
def on_first_page(canvas_obj, doc):
on_page(canvas_obj, doc)
# ── Styles ───────────────────────────────────────────────────────────
styles = getSampleStyleSheet()
title_style = ParagraphStyle(
"DocTitle",
parent=styles["Title"],
fontSize=26,
textColor=WHITE,
alignment=TA_CENTER,
spaceAfter=4,
fontName="Helvetica-Bold",
)
subtitle_style = ParagraphStyle(
"DocSubtitle",
parent=styles["Normal"],
fontSize=11,
textColor=HexColor("#FADBD8"),
alignment=TA_CENTER,
spaceAfter=2,
fontName="Helvetica",
)
source_style = ParagraphStyle(
"Source",
parent=styles["Normal"],
fontSize=8,
textColor=HexColor("#FADBD8"),
alignment=TA_CENTER,
fontName="Helvetica-Oblique",
)
h1 = ParagraphStyle(
"H1",
parent=styles["Heading1"],
fontSize=14,
textColor=WHITE,
fontName="Helvetica-Bold",
spaceBefore=14,
spaceAfter=6,
leftIndent=0,
borderPad=6,
)
h2 = ParagraphStyle(
"H2",
parent=styles["Heading2"],
fontSize=11,
textColor=DEEP_RED,
fontName="Helvetica-Bold",
spaceBefore=10,
spaceAfter=4,
borderColor=DEEP_RED,
)
body = ParagraphStyle(
"Body",
parent=styles["Normal"],
fontSize=9.5,
textColor=DARK_GREY,
fontName="Helvetica",
spaceAfter=5,
leading=14,
alignment=TA_JUSTIFY,
)
bullet = ParagraphStyle(
"Bullet",
parent=body,
leftIndent=14,
bulletIndent=4,
spaceBefore=1,
spaceAfter=3,
)
caption = ParagraphStyle(
"Caption",
parent=styles["Normal"],
fontSize=8,
textColor=HexColor("#566573"),
fontName="Helvetica-Oblique",
alignment=TA_CENTER,
spaceAfter=8,
spaceBefore=3,
)
small = ParagraphStyle(
"Small",
parent=body,
fontSize=8,
textColor=HexColor("#566573"),
)
# ── Helper: coloured section header banner ────────────────────────────
def section_header(text, bg=DEEP_RED):
data = [[Paragraph(f"<font color='white'><b>{text}</b></font>", h1)]]
t = Table(data, colWidths=[17.5*cm])
t.setStyle(TableStyle([
("BACKGROUND", (0,0), (-1,-1), bg),
("TOPPADDING", (0,0), (-1,-1), 7),
("BOTTOMPADDING", (0,0), (-1,-1), 7),
("LEFTPADDING", (0,0), (-1,-1), 10),
("RIGHTPADDING", (0,0), (-1,-1), 10),
("ROUNDEDCORNERS", [4]),
]))
return t
# ── Helper: two-column info box ───────────────────────────────────────
def info_table(rows, col_widths=(6*cm, 11.5*cm)):
data = []
for label, value in rows:
data.append([
Paragraph(f"<b>{label}</b>", small),
Paragraph(value, small),
])
t = Table(data, colWidths=col_widths)
t.setStyle(TableStyle([
("BACKGROUND", (0, 0), (0, -1), PALE_GREY),
("BACKGROUND", (1, 0), (1, -1), WHITE),
("GRID", (0, 0), (-1, -1), 0.4, MID_GREY),
("TOPPADDING", (0, 0), (-1, -1), 4),
("BOTTOMPADDING", (0, 0), (-1, -1), 4),
("LEFTPADDING", (0, 0), (-1, -1), 6),
("VALIGN", (0, 0), (-1, -1), "TOP"),
]))
return t
# ── Helper: styled data table ─────────────────────────────────────────
def data_table(headers, rows, col_widths=None):
header_row = [Paragraph(f"<b><font color='white'>{h}</font></b>", small) for h in headers]
body_rows = [[Paragraph(str(c), small) for c in row] for row in rows]
all_rows = [header_row] + body_rows
if col_widths is None:
col_widths = [17.5*cm / len(headers)] * len(headers)
t = Table(all_rows, colWidths=col_widths, repeatRows=1)
t.setStyle(TableStyle([
("BACKGROUND", (0, 0), (-1, 0), DEEP_RED),
("BACKGROUND", (0, 1), (-1, -1), WHITE),
("ROWBACKGROUNDS",(0, 1), (-1, -1), [WHITE, PALE_GREY]),
("GRID", (0, 0), (-1, -1), 0.4, MID_GREY),
("TOPPADDING", (0, 0), (-1, -1), 4),
("BOTTOMPADDING", (0, 0), (-1, -1), 4),
("LEFTPADDING", (0, 0), (-1, -1), 5),
("VALIGN", (0, 0), (-1, -1), "TOP"),
("FONTSIZE", (0, 0), (-1, -1), 8.5),
]))
return t
# ── Helper: highlight box ────────────────────────────────────────────
def highlight_box(text, bg=LIGHT_RED, border=DARK_RED):
data = [[Paragraph(text, ParagraphStyle("hb", parent=body, fontSize=9, spaceAfter=0))]]
t = Table(data, colWidths=[17.5*cm])
t.setStyle(TableStyle([
("BACKGROUND", (0,0), (-1,-1), bg),
("TOPPADDING", (0,0), (-1,-1), 8),
("BOTTOMPADDING", (0,0), (-1,-1), 8),
("LEFTPADDING", (0,0), (-1,-1), 12),
("RIGHTPADDING", (0,0), (-1,-1), 12),
("LINEAFTER", (0,0), (0,-1), 4, border),
("BOX", (0,0), (-1,-1), 0.5, border),
]))
return t
# ════════════════════════════════════════════════════════════════════
# BUILD STORY
# ════════════════════════════════════════════════════════════════════
story = []
W, H = A4
# ── COVER BANNER ────────────────────────────────────────────────────
cover_data = [[
Paragraph("Berry (Saccular) Aneurysm", title_style),
Paragraph("Pathology & Morphology – Summary Reference", subtitle_style),
Paragraph("Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease | Harrison's Principles of Internal Medicine 22e | Neuroanatomy through Clinical Cases 3e", source_style),
]]
cover = Table(cover_data, colWidths=[17.5*cm])
cover.setStyle(TableStyle([
("BACKGROUND", (0,0), (-1,-1), DEEP_RED),
("TOPPADDING", (0,0), (-1,-1), 22),
("BOTTOMPADDING", (0,0), (-1,-1), 22),
("LEFTPADDING", (0,0), (-1,-1), 16),
("RIGHTPADDING", (0,0), (-1,-1), 16),
("ROUNDEDCORNERS", [6]),
]))
story.append(cover)
story.append(Spacer(1, 0.5*cm))
# ── 1. DEFINITION ───────────────────────────────────────────────────
story.append(section_header("1. Definition"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph(
"A <b>berry (saccular) aneurysm</b> is a thin-walled, round or lobulated outpouching arising at the "
"<b>bifurcation points of intracranial arteries</b>, most commonly those forming the Circle of Willis. "
"It is the <b>most common type of intracranial aneurysm</b> and the leading cause of spontaneous "
"subarachnoid haemorrhage (SAH). The term 'berry' refers to its rounded, berry-like gross appearance.",
body
))
story.append(Spacer(1, 0.3*cm))
# ── 2. EPIDEMIOLOGY ─────────────────────────────────────────────────
story.append(section_header("2. Epidemiology"))
story.append(Spacer(1, 0.2*cm))
story.append(info_table([
("Prevalence", "~2% of the adult population; ~4 million persons in the United States"),
("SAH incidence", "6–11 per 100,000 person-years; ~25,000–30,000 cases/year in the US"),
("Peak age", "Fifth decade (40s–50s); incidence increases with age"),
("Sex", "Slightly more frequent in females (1.3× relative risk)"),
("Multiple aneurysms", "20–30% of cases have multiple aneurysms, many at mirror sites bilaterally"),
("Location", "~90% in anterior circulation; 89% on or near Circle of Willis"),
("Mortality (1st rupture)", "25–50% die immediately or before reaching hospital; overall SAH mortality ~35%"),
]))
story.append(Spacer(1, 0.4*cm))
# ── 3. SITES / FREQUENCY ────────────────────────────────────────────
story.append(section_header("3. Common Sites of Occurrence"))
story.append(Spacer(1, 0.2*cm))
# two columns: table left, image right
img_path = "/home/daytona/workspace/berry-aneurysm/aneurysm_sites.png"
site_rows = [
["Anterior communicating artery (AComm)", "38% – Most common"],
["Anterior cerebral artery (ACA)", "36%"],
["Middle cerebral artery (MCA) bifurcation","21%"],
["Basilar artery / vertebrobasilar", "5%"],
["Posterior communicating artery (PComm)", "Clinically important – CN III palsy"],
["Internal carotid artery (ICA) terminus", "Giant aneurysms common here"],
]
site_table = data_table(
["Location", "Frequency / Notes"],
site_rows,
col_widths=[8.5*cm, 5*cm]
)
img_w = 4*cm
try:
from PIL import Image as PILImage
pil = PILImage.open(img_path)
orig_w, orig_h = pil.size
img_h = img_w * orig_h / orig_w
except Exception:
img_h = 5*cm
site_img = Image(img_path, width=img_w, height=img_h) if os.path.exists(img_path) else Spacer(1,1)
col_layout = Table(
[[site_table, site_img]],
colWidths=[13.5*cm, 4*cm]
)
col_layout.setStyle(TableStyle([
("VALIGN", (0,0), (-1,-1), "TOP"),
("LEFTPADDING", (0,0), (-1,-1), 0),
("RIGHTPADDING", (0,0), (-1,-1), 0),
("TOPPADDING", (0,0), (-1,-1), 0),
]))
story.append(col_layout)
story.append(Paragraph(
"Fig. 1. Common sites of saccular (berry) aneurysm in the Circle of Willis "
"(Bailey & Love; Robbins PBD Fig. 28.15).",
caption
))
# ── 4. PATHOGENESIS ─────────────────────────────────────────────────
story.append(section_header("4. Pathogenesis"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph("<b>Structural Wall Defect:</b>", h2))
story.append(Paragraph(
"Saccular aneurysms are not present at birth but develop over time due to an <b>underlying defect "
"in the tunica media</b> of the vessel wall. The arterial internal elastic lamina <b>disappears at the "
"base of the neck</b>. The media thins and smooth-muscle cells are replaced by connective tissue. "
"At the site of rupture (usually the dome), the wall is at its thinnest and the tear "
"allowing bleeding is often ≤0.5 mm long.",
body
))
story.append(Paragraph("<b>Haemodynamic Stress:</b>", h2))
story.append(Paragraph(
"Turbulent blood flow at arterial bifurcation points creates focal endothelial dysfunction. "
"Over time this progressive haemodynamic stress, combined with the structural wall defect, "
"promotes outpouching and enlargement. <b>Hypertension</b> (present in ~50% of affected individuals) "
"and <b>smoking</b> are major acquired risk factors that accelerate this process.",
body
))
story.append(Paragraph("<b>Molecular / Genetic Factors:</b>", h2))
story.append(Paragraph(
"The majority of saccular aneurysms occur sporadically. However, genetic factors are important: "
"there is an increased incidence in first-degree relatives of affected individuals, and a "
"significantly higher incidence in the following Mendelian disorders:",
body
))
genetic_rows = [
["Autosomal dominant polycystic kidney disease (ADPKD)", "Most strongly associated; connective tissue defect in arterial walls"],
["Ehlers-Danlos syndrome type IV", "Defective Type III collagen"],
["Marfan syndrome", "Fibrillin-1 mutation; connective tissue weakness"],
["Neurofibromatosis type 1 (NF1)", "Vascular dysplasia"],
["Fibromuscular dysplasia", "Non-inflammatory arterial disease"],
["Coarctation of the aorta", "Altered haemodynamics and hypertension"],
]
story.append(data_table(
["Associated Condition", "Mechanism"],
genetic_rows,
col_widths=[8*cm, 9.5*cm]
))
story.append(Spacer(1, 0.3*cm))
story.append(highlight_box(
"<b>Key Point:</b> Although called 'congenital', saccular aneurysms are NOT present at birth. "
"They develop over decades from an underlying medial defect combined with haemodynamic wear at bifurcation points."
))
story.append(Spacer(1, 0.4*cm))
# ── 5. MORPHOLOGY ──────────────────────────────────────────────────
story.append(section_header("5. Morphology"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph("<b>Gross Appearance:</b>", h2))
morph_rows = [
("Shape", "Round or lobulated thin-walled outpouching; 'berry-like'"),
("Size", "Few mm to 2–3 cm in diameter. Giant aneurysms >25 mm (5% of cases)"),
("Surface", "Bright red, shiny, translucent wall"),
("Location", "Arise at arterial branch point; have a neck (narrow or wide) and a dome"),
("Neck", "Site where aneurysm meets parent artery; no media or elastic lamina"),
("Dome", "Thinnest part; site of rupture; tear usually ≤0.5 mm"),
("Contents", "Atheromatous plaques, calcification, or mural thrombi may be present in wall/lumen"),
("Prior bleeds","Orange-brown discolouration of adjacent brain and meninges"),
]
story.append(info_table(morph_rows))
story.append(Spacer(1, 0.3*cm))
# Gross image
img2_path = "/home/daytona/workspace/berry-aneurysm/berry_gross.png"
if os.path.exists(img2_path):
try:
from PIL import Image as PILImage
pil2 = PILImage.open(img2_path)
o2w, o2h = pil2.size
iw2 = 14*cm
ih2 = iw2 * o2h / o2w
except Exception:
iw2, ih2 = 14*cm, 5*cm
story.append(Image(img2_path, width=iw2, height=ih2))
story.append(Paragraph(
"Fig. 2. Berry aneurysm – gross and microscopic morphology. "
"(A) Base of brain with aneurysm of ACA (arrow). "
"(B) Dissected Circle of Willis showing large aneurysm. "
"(C) Histology: hyalinized fibrous wall (H&E). "
"Source: Robbins, Cotran & Kumar PBD, Fig. 28.16.",
caption
))
story.append(Paragraph("<b>Histological Appearance:</b>", h2))
histo_rows = [
("Normal artery wall", "Intima + Internal elastic lamina + Tunica media (smooth muscle) + Adventitia"),
("Aneurysm neck", "Intimal thickening; media attenuated; elastic lamina absent"),
("Aneurysm sac (dome)", "Thickened hyalinized intima + adventitia ONLY. No media. No elastic lamina."),
("Rupture site", "Wall thinning to near transparency; tear ≤0.5 mm at dome apex"),
("Secondary changes", "Intraluminal thrombus; atheromatous change; calcification in chronic cases"),
]
story.append(info_table(histo_rows))
story.append(Spacer(1, 0.3*cm))
story.append(highlight_box(
"<b>Histological Key:</b> The aneurysm sac wall consists of <b>thickened hyalinized intima</b> "
"covered only by <b>adventitia</b>. There is a complete <b>absence of smooth muscle and internal "
"elastic lamina</b> – this distinguishes it from a normal artery or atherosclerotic fusiform aneurysm."
))
story.append(Spacer(1, 0.4*cm))
# ── 6. RUPTURE AND SAH ─────────────────────────────────────────────
story.append(section_header("6. Rupture and Subarachnoid Haemorrhage (SAH)"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph("<b>Risk of Rupture:</b>", h2))
rupture_rows = [
["Aneurysm <3 mm", "Rarely bleeds; risk essentially 0% at 5 years"],
["Aneurysm <7 mm", "Very low annual risk in most locations"],
["Aneurysm 7–10 mm", "Intermediate risk; surveillance vs. treatment decision"],
["Aneurysm >10 mm", "~50% risk per year; treatment strongly recommended"],
["Giant >25 mm", "8–10% annual rupture risk; may also cause mass effect"],
["Overall (all sizes)","1.3% per year"],
]
story.append(data_table(
["Size", "Rupture Risk"],
rupture_rows,
col_widths=[5*cm, 12.5*cm]
))
story.append(Spacer(1, 0.3*cm))
story.append(Paragraph("<b>Triggers of Rupture:</b>", h2))
for item in [
"Acute rises in intracranial pressure: straining at stool, sexual orgasm, heavy lifting (~1/3 of cases)",
"Physical exertion or Valsalva manoeuvre",
"Spontaneous rupture during sleep or rest",
"Uncontrolled hypertension",
]:
story.append(Paragraph(f"• {item}", bullet))
story.append(Spacer(1, 0.3*cm))
story.append(Paragraph("<b>Consequences of Rupture:</b>", h2))
story.append(Paragraph(
"Blood under <b>arterial pressure</b> is forced into the subarachnoid space (basal cisterns) "
"and sometimes into brain parenchyma. This causes sudden, severe intracranial hypertension "
"and the characteristic clinical picture.",
body
))
conseq_rows = [
["Acute SAH", "Blood fills subarachnoid space; basal cisterns packed with clot"],
["Intracerebral haematoma", "Blood may track into parenchyma adjacent to aneurysm"],
["Intraventricular haemorrhage","Blood enters ventricles; worsens prognosis"],
["Brain herniation", "Acute rise in ICP may cause transtentorial herniation; death"],
["Vasospasm (days 4–14)", "Narrowing of Circle of Willis arteries → cerebral ischaemia (30% of survivors)"],
["Hydrocephalus", "Acute (CSF obstruction) or chronic (meningeal scarring)"],
["Hyponatremia", "Cerebral salt wasting; first 2 weeks"],
]
story.append(data_table(
["Complication", "Details"],
conseq_rows,
col_widths=[5.5*cm, 12*cm]
))
story.append(Spacer(1, 0.4*cm))
# ── 7. CLINICAL FEATURES ──────────────────────────────────────────
story.append(section_header("7. Clinical Features"))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph("<b>Presentation of Ruptured Aneurysm:</b>", h2))
for item in [
'<b>Thunderclap headache</b> – sudden, excruciating ("worst headache of my life")',
"<b>Loss of consciousness</b> in ~50% of cases",
"<b>Vomiting</b> in ~70% of cases",
"<b>Seizure</b> in ~10% of cases",
"<b>Meningism</b> (neck stiffness, photophobia) – develops over hours",
"<b>Sentinel bleed</b> – sudden unexplained headache days before major rupture; must not be missed",
]:
story.append(Paragraph(f"• {item}", bullet))
story.append(Spacer(1, 0.2*cm))
story.append(Paragraph("<b>Location-Specific Signs:</b>", h2))
location_rows = [
["Posterior communicating artery (PComm)", "Painful 3rd nerve palsy (CN III) – ptosis, dilated pupil, eye 'down and out'"],
["Anterior communicating artery (AComm)", "Abulia, leg weakness, memory disturbance"],
["Middle cerebral artery (MCA)", "Contralateral hemiplegia + aphasia (dominant side)"],
["Basilar artery tip", "Altered consciousness; 3rd nerve palsy; top-of-basilar syndrome"],
["Internal carotid artery", "Visual field defects; unilateral blindness if ophthalmic involved"],
]
story.append(data_table(
["Aneurysm Site", "Clinical Sign"],
location_rows,
col_widths=[6.5*cm, 11*cm]
))
story.append(Spacer(1, 0.3*cm))
story.append(Paragraph("<b>Grading Scales (Hunt-Hess):</b>", h2))
hh_rows = [
["Grade 1", "Asymptomatic or mild headache", "Excellent"],
["Grade 2", "Moderate to severe headache; nuchal rigidity; no deficit except CN palsy", "Good"],
["Grade 3", "Drowsiness, confusion, mild focal deficit", "Fair"],
["Grade 4", "Stupor, moderate to severe hemiparesis", "Poor (mortality ~60%)"],
["Grade 5", "Deep coma, decerebrate rigidity, moribund", "Very poor"],
]
story.append(data_table(
["Grade", "Description", "Prognosis"],
hh_rows,
col_widths=[2.5*cm, 10*cm, 5*cm]
))
story.append(Spacer(1, 0.4*cm))
# ── 8. TYPES OF INTRACRANIAL ANEURYSM ─────────────────────────────
story.append(section_header("8. Types of Intracranial Aneurysm – Comparison"))
story.append(Spacer(1, 0.2*cm))
types_rows = [
["Saccular (berry)", "Circle of Willis bifurcations", "Most common; subarachnoid haemorrhage", "Congenital medial defect + haemodynamic stress"],
["Fusiform (atherosclerotic)", "Basilar artery (mostly)", "Ischaemic stroke; mass effect", "Atherosclerosis; hypertension"],
["Mycotic", "Distal to 1st bifurcation of CoW vessels", "Haemorrhage; infarction", "Septic emboli; infective endocarditis"],
["Traumatic", "Peripheral branches", "Delayed haemorrhage", "Direct injury or shear forces"],
["Dissecting", "Any intracranial vessel", "Ischaemic or haemorrhagic stroke", "Intimal tear; blood in vessel wall"],
]
story.append(data_table(
["Type", "Location", "Main Complication", "Mechanism"],
types_rows,
col_widths=[3.5*cm, 4.5*cm, 4.5*cm, 5*cm]
))
story.append(Spacer(1, 0.4*cm))
# ── 9. INVESTIGATIONS ─────────────────────────────────────────────
story.append(section_header("9. Investigations"))
story.append(Spacer(1, 0.2*cm))
inv_rows = [
("Non-contrast CT brain (immediate)", "Hyperdense blood in subarachnoid space; sensitivity >95% within 6 hours of SAH"),
("Lumbar puncture (if CT negative)", "Xanthochromia (yellow CSF due to bilirubin from haemoglobin breakdown) after 12 hours; highly sensitive"),
("CT angiography (CTA)", "Non-invasive; detects aneurysms >3 mm; images Circle of Willis; vasospasm assessment"),
("Digital subtraction angiography (DSA)","Gold standard; shows aneurysm anatomy (neck, dome, parent vessel); guides treatment planning"),
("MRI / MRA", "Useful for unruptured aneurysm detection; less sensitive than DSA for small lesions"),
("Transcranial Doppler (TCD)", "Daily monitoring for vasospasm; detects elevated MCA/ACA flow velocities"),
]
story.append(info_table(inv_rows, col_widths=[5.5*cm, 12*cm]))
story.append(Spacer(1, 0.4*cm))
# ── 10. TREATMENT ─────────────────────────────────────────────────
story.append(section_header("10. Treatment"))
story.append(Spacer(1, 0.2*cm))
treat_rows = [
["Surgical clipping", "Gold standard for anterior circulation aneurysms; metallic clip across neck", "Immediate exclusion; durable", "Craniotomy required; higher surgical risk in poor-grade patients"],
["Endovascular coiling (GDC)", "Platinum coils packed into aneurysm dome via catheter", "Less invasive; suitable for posterior circulation", "Higher rebleed rate long-term; may need re-treatment"],
["Flow diversion (pipeline embolization)", "Stent redirects flow away from aneurysm", "Giant or fusiform aneurysms", "Delayed occlusion; antiplatelet therapy required"],
["Nimodipine (calcium channel blocker)", "Pharmacological prevention of vasospasm", "All SAH patients", "Does not reduce vasospasm but improves neurological outcome"],
["Triple-H therapy (historical)", "Hypertension + Hypervolaemia + Haemodilution", "Symptomatic vasospasm", "Now replaced by euvolaemia + BP management"],
]
story.append(data_table(
["Modality", "Description", "Best For", "Limitation"],
treat_rows,
col_widths=[4*cm, 5.5*cm, 3.5*cm, 4.5*cm]
))
story.append(Spacer(1, 0.4*cm))
# ── 11. KEY FACTS BOX ─────────────────────────────────────────────
story.append(section_header("11. Examination Key Points"))
story.append(Spacer(1, 0.2*cm))
kf_data = [
["Most common type of intracranial aneurysm", "Saccular (berry) aneurysm"],
["Prevalence in general population", "~2%"],
["Anterior circulation involvement", "~90%"],
["Most common single site", "Anterior communicating artery (AComm) – 38%"],
["Classic presentation", "Thunderclap headache ('worst of my life')"],
["CN palsy associated with PComm aneurysm", "Oculomotor (CN III) palsy – dilated pupil, ptosis, 'down and out' eye"],
["Key histological finding", "No smooth muscle, no elastic lamina in sac wall – hyalinized intima + adventitia only"],
["Vasospasm peak", "Day 4–14 post-SAH"],
["Vasospasm treatment", "Nimodipine (oral); endovascular balloon/chemical angioplasty"],
["Rebleed risk (untreated)", "~30% in first month; 50% mortality with rebleed"],
["Overall mortality with first rupture", "25–50%"],
["Genetic associations", "ADPKD, Ehlers-Danlos IV, Marfan, NF1, coarctation of aorta"],
]
kf_table = Table(
[[Paragraph(f"<b>{r[0]}</b>", small), Paragraph(r[1], small)] for r in kf_data],
colWidths=[8*cm, 9.5*cm]
)
kf_table.setStyle(TableStyle([
("ROWBACKGROUNDS", (0,0), (-1,-1), [PALE_GREY, WHITE]),
("GRID", (0,0), (-1,-1), 0.4, MID_GREY),
("TOPPADDING", (0,0), (-1,-1), 4),
("BOTTOMPADDING", (0,0), (-1,-1), 4),
("LEFTPADDING", (0,0), (-1,-1), 6),
("VALIGN", (0,0), (-1,-1), "TOP"),
]))
story.append(kf_table)
story.append(Spacer(1, 0.4*cm))
# ── REFERENCE FOOTER ─────────────────────────────────────────────
story.append(HRFlowable(width="100%", thickness=1, color=MID_GREY, spaceAfter=6))
story.append(Paragraph(
"<b>References:</b> (1) Robbins, Cotran & Kumar – Pathologic Basis of Disease, 10th Ed., "
"ISBN 9780443264528, pp. 1157–1159. "
"(2) Harrison's Principles of Internal Medicine, 22nd Ed. (2025), ISBN 9781265977061, Chapter 440. "
"(3) Neuroanatomy through Clinical Cases, 3rd Ed., ISBN 9781605359625, p. 417. "
"(4) Bailey & Love's Short Practice of Surgery, 28th Ed., Chapter 48.",
ParagraphStyle("ref", parent=small, fontSize=7.5, textColor=HexColor("#7F8C8D"))
))
# ════════════════════════════════════════════════════════════════════
# RENDER PDF
# ════════════════════════════════════════════════════════════════════
doc = SimpleDocTemplate(
OUTPUT,
pagesize=A4,
topMargin=1.6*cm,
bottomMargin=1.8*cm,
leftMargin=1.5*cm,
rightMargin=1.5*cm,
title="Berry Aneurysm – Pathology & Morphology Summary",
author="Orris Medical AI",
subject="Berry Aneurysm Pathology",
)
doc.build(story, onFirstPage=on_first_page, onLaterPages=on_page)
print(f"PDF generated: {OUTPUT}")
cd /home/daytona/workspace/berry-aneurysm && python generate_pdf.py 2>&1
ls -lh /home/daytona/workspace/berry-aneurysm/Berry_Aneurysm_Summary.pdf
Berry Aneurysm Summary
PDF Document · PDF
- Definition - concise clinical definition
- Epidemiology - prevalence, incidence, sex ratio, mortality data
- Common Sites - frequency table alongside the Circle of Willis aneurysm site diagram
- Pathogenesis - structural wall defect, haemodynamic stress, genetic/Mendelian associations table
- Morphology - gross appearance (with the Robbins gross/histology image), full histological description table, and a highlighted key facts box
- Rupture & SAH - rupture risk by size, triggers, complications table (vasospasm, hydrocephalus, hyponatremia)
- Clinical Features - thunderclap headache, location-specific signs (CN III palsy), Hunt-Hess grading table
- Types of Intracranial Aneurysm - comparison table (saccular vs. fusiform vs. mycotic vs. traumatic)
- Investigations - CT, LP, CTA, DSA, TCD
- Treatment - clipping vs. coiling vs. flow diversion; nimodipine
- Examination Key Points - quick-reference table of the most exam-tested facts
Pathophysiology of Organophosphate poisoning , with is clinical features, management, complication, add forensic medicine aspect as this is for pg FMT paper
Organophosphate (OP) Poisoning
Pathophysiology, Clinical Features, Management, Complications & Forensic Aspects
1. Introduction
- Agricultural insecticides (malathion, parathion, chlorpyrifos, diazinon, acephate)
- Chemical warfare nerve agents (sarin, soman, tabun, VX)
- Veterinary preparations (flea dips, anthelmintics)
2. Pathophysiology
2A. Mechanism of Toxicity
- Muscarinic receptors (parasympathetic end-organs, sweat glands)
- Nicotinic receptors (neuromuscular junction, autonomic ganglia, adrenal medulla)
- CNS cholinergic synapses
2B. Concept of "Aging"
- Aging = the permanent, irreversible binding of the OP compound to AChE after initial phosphorylation
- Once aging occurs, enzymatic activity is permanently destroyed - new AChE must be resynthesized over weeks
- Time to aging is highly variable - from minutes (soman - within minutes; critical for nerve agent antidote window) to days (malathion)
- Pralidoxime (oxime antidote) MUST be given before aging occurs - it is ineffective once aging is complete
2C. Two Cholinesterase Enzymes Affected
| Enzyme | Location | Role |
|---|---|---|
| Acetylcholinesterase (AChE) - "true" or RBC cholinesterase | Erythrocyte membranes, nervous tissue, skeletal muscle | Primary site of OP action; more accurate indicator of synaptic inhibition |
| Butyrylcholinesterase (BChE) - pseudocholinesterase | Serum, liver, pancreas, heart, brain | Used for lab monitoring; easier to assay but less specific |
2D. Three Receptor Sites Involved
| Receptor Type | Location | Effect of ACh Excess |
|---|---|---|
| Muscarinic (M1-M5) | Smooth muscle, secretory glands, cardiac SA/AV nodes, eye | SLUDGE/DUMBELS symptoms |
| Nicotinic (NMJ) | Neuromuscular junction (skeletal muscle) | Fasciculations → paralysis |
| Nicotinic (ganglionic) | Autonomic ganglia, adrenal medulla | Sympathetic stimulation: tachycardia, hypertension, mydriasis |
| CNS | Entire brain | Anxiety → seizures → coma |
3. Routes of Exposure
| Route | Onset | Common Scenario |
|---|---|---|
| Inhalation | Minutes (fastest) | Pesticide spray, nerve agents, industrial accident |
| Ingestion | Minutes to hours | Deliberate self-poisoning (most common globally), accidental |
| Dermal/transconjunctival | Hours (slowest - but enhanced by skin excoriation) | Agricultural workers, flea dip products |
| Mucous membrane | Intermediate | Spray drift |
4. Clinical Features
4A. Four Clinical Syndromes
| Syndrome | Onset | Features |
|---|---|---|
| Acute cholinergic toxidrome | Minutes to 24 hours | Full muscarinic + nicotinic + CNS effects |
| Intermediate syndrome | 1-5 days post-acute phase | Proximal muscle + respiratory paralysis without cholinergic excess |
| Organophosphate-induced delayed neuropathy (OPIDN) | 2-5 weeks post-exposure | Distal sensorimotor polyneuropathy |
| Chronic toxicity | Ongoing occupational exposure | Cognitive, autonomic, neuropsychiatric effects |
4B. Acute Cholinergic Toxidrome
MUSCARINIC EFFECTS - Mnemonic: SLUDGE / DUMBELS / "Killer Bs"
| Mnemonic | Feature |
|---|---|
| S | Salivation (excessive) |
| L | Lacrimation |
| U | Urinary incontinence |
| D | Defecation (diarrhoea) |
| G | GI cramping / pain |
| E | Emesis (vomiting) |
| Mnemonic | Feature |
|---|---|
| D | Defecation |
| U | Urination |
| M | Miosis (pinpoint pupils - hallmark sign) |
| B | Bradycardia |
| E | Emesis |
| L | Lacrimation |
| S | Salivation, Sweating |
- Bradycardia
- Bronchorrhea (profuse watery secretions)
- Bronchospasm
- Increased peristalsis
- Urinary/faecal incontinence
- Diaphoresis (sweating)
- Miosis - most consistent and diagnostically important sign
NICOTINIC EFFECTS (Neuromuscular Junction)
- Muscle fasciculations - visible twitching, pathognomonic
- Muscle cramps
- Progressive muscle weakness
- Respiratory muscle paralysis (diaphragm + intercostals) → ventilatory failure
- Areflexia in severe cases (masking seizure activity)
NICOTINIC EFFECTS (Ganglionic - Sympathetic)
- Tachycardia (may override bradycardia)
- Hypertension
- Mydriasis (may counteract or coexist with miosis)
- Pallor
Note: Mixed autonomic effects are common. Parasympathetic usually predominates, but sympathetic "nicotinic escape" occurs in severe poisoning.
CNS EFFECTS
| Severity | Features |
|---|---|
| Mild | Anxiety, restlessness, emotional lability |
| Moderate | Tremor, headache, dizziness, confusion, delirium, hallucinations |
| Severe | Seizures, coma |
| Terminal | Respiratory centre depression |
- Altered mental status
- Pinpoint pupils (miosis)
- Excessive sweating
- Difficulty breathing
4C. Intermediate Syndrome (IMS)
- Occurs 1-5 days after the acute cholinergic phase (up to 40% of ingestion cases)
- Signs of cholinergic excess are absent
- Features:
- Paralysis of neck flexors
- Paralysis of proximal limb muscles
- Cranial nerve motor palsies
- Respiratory muscle paralysis → ventilatory failure → death if unrecognised
- EMG assists diagnosis
- Responds to respiratory support ONLY (not atropine/pralidoxime)
- Resolves in 7-21 days
4D. Organophosphate-Induced Delayed Neuropathy (OPIDN)
- Appears 2-5 weeks after acute exposure (classic with TOCP - triorthocresyl phosphate)
- Distal symmetrical sensorimotor polyneuropathy (predominantly motor)
- Begins with leg cramps → weakness → ataxia → paralysis
- Mimics Guillain-Barré syndrome
- Mechanism: inhibition of neuropathy target esterase (NTE) → "dying-back" axonopathy from terminal ends of longest motor fibres
- Late finding: corticospinal tract signs (in TOCP poisoning)
- No effective treatment - supportive only; variable recovery
5. Diagnosis
Clinical Diagnosis
- Based on history + cholinergic toxidrome
- Characteristic garlic/hydrocarbon odour
- Do NOT wait for lab results before treating
Laboratory Tests
| Test | Significance |
|---|---|
| Red cell (true) AChE activity | Most accurate indicator of synaptic inhibition; correlates with clinical severity |
| Plasma BChE (pseudocholinesterase) | More available; falls earlier; >50% inhibition = significant exposure |
| ABG | Hypoxaemia, respiratory acidosis |
| ECG | QTc prolongation, bradycardia, heart block, ventricular arrhythmias |
| Blood glucose | Hyperglycaemia common |
| Serum electrolytes | Hypokalaemia |
| Chest X-ray | Aspiration pneumonia, pulmonary oedema |
Baseline cholinesterase activity varies greatly among individuals; single values have limited utility without serial monitoring.
6. Management
Step 1 - Decontamination (Priority: Protect Rescuers First)
- PPE for healthcare workers: Full-face air purifier mask, chemical-resistant suit, nitrile/butyl rubber gloves, eye shield (Level C PPE)
- Remove and destroy all clothing
- Thorough skin flushing with water (primary decontamination method)
- Equipment may be decontaminated with 5% hypochlorite solution
- Gastric lavage and activated charcoal are NOT recommended - OP is rapidly absorbed; profuse vomiting occurs early; risk of aspiration
Step 2 - Airway and Supportive Care
- Suction secretions and vomitus
- Supplemental oxygen
- Endotracheal intubation and mechanical ventilation if required
- Use non-depolarizing paralytic (rocuronium 1 mg/kg) for RSI, NOT succinylcholine
- Succinylcholine is metabolized by AChE - prolonged paralysis (4-6 hours) occurs in OP poisoning
- Benzodiazepines for seizures and agitation (after airway secured)
- Do NOT treat tachycardia with beta-blockers - resolves with atropine
Step 3 - Antidote 1: ATROPINE
- Adults: 1-3 mg IV bolus initially; double the dose every 5 minutes until muscarinic symptoms controlled
- Children: 0.05 mg/kg IV
- Total doses up to 200-500 mg in the first hour may be required in severe cases
- Once stabilised: maintenance infusion of 10-20% of total loading dose per hour
- Drying of respiratory secretions
- Easing of respiratory effort
- Normalisation of respiratory rate
- Clearing of bronchospasm
Tachycardia and mydriasis at high atropine doses are NOT indications to stop - they are expected. Endpoint is drying of secretions.
Step 4 - Antidote 2: PRALIDOXIME (2-PAM / Oxime)
- Respiratory depression or failure
- Muscle fasciculations
- Seizures
- Dysrhythmias
- Haemodynamic instability
- Requirement for large/repeated atropine doses
- 1-2 g IV over 30 minutes (25-50 mg/kg in children); can repeat hourly based on response
- Alternative: 2 g bolus then infusion 500 mg/h for up to 7 days
Step 5 - Seizure Management
- Benzodiazepines (diazepam/lorazepam) - first line
- Seizures may be occult (paralysed patient; areflexia masks convulsions)
- Monitor with EEG if intubated and paralysed
Step 6 - Disposition
| Presentation | Disposition |
|---|---|
| Asymptomatic / mild, normal or minimal cholinesterase depression | Observe 6 hours; discharge with close follow-up |
| Significant symptoms (seizures, respiratory compromise) | ICU admission; continuous monitoring |
| All significant exposures | Admit to monitored setting; rebound toxicity possible days later |
7. Complications
| Complication | Mechanism | Timing |
|---|---|---|
| Respiratory failure | Bronchorrhea + bronchospasm + respiratory muscle paralysis | Acute - leading cause of death |
| Aspiration pneumonia / lipoid pneumonia | Aspiration of vomitus and hydrocarbon solvents | Acute |
| Cardiac arrhythmias | QTc prolongation, bradycardia, torsades de pointes, ventricular fibrillation | Acute |
| Myocardial ischaemia | Cholinergic crisis affecting coronary blood flow | Acute |
| Seizures / Status epilepticus | CNS cholinergic excess | Acute |
| Acute pancreatitis | Direct OP effect on pancreatic cells | Acute |
| Intermediate syndrome (IMS) | NMJ dysfunction post-cholinergic phase | Days 1-5 |
| OPIDN | Neuropathy target esterase (NTE) inhibition | 2-5 weeks |
| Chronic neuropsychiatric effects | Low-level AChE inhibition; oxidative stress | Chronic exposure |
| Cognitive impairment / PTSD | CNS damage from hypoxia + direct OP toxicity | Long-term |
| Corticospinal tract damage (TOCP) | Dying-back axonopathy in spinal cord | Weeks-months after TOCP |
8. Forensic Medicine Aspects (FMT - Key Points)
8A. Medicolegal Significance
- OP poisoning is the single most common cause of suicidal death by poisoning in India and many developing countries
- Also accounts for accidental deaths (farm workers, children) and homicidal deaths (rare, but documented)
- A notifiable condition in many jurisdictions
8B. Mode of Death / Manner of Death
| Manner | Circumstances |
|---|---|
| Suicide | Most common; deliberate ingestion; young adults in rural agricultural communities |
| Accidental | Occupational (farm workers, pesticide applicators); children (flea dip products, contaminated food); inadvertent inhalation |
| Homicidal | Rare; OP compounds added to food or drink; witnessed administration; forensic toxicology required |
| Natural | Not applicable |
8C. Autopsy Findings (Post-mortem)
- Miosis (pinpoint pupils) - most consistent and important external finding
- Excessive secretions: froth from mouth/nose (profuse bronchial secretions)
- Garlic/hydrocarbon odour from body and viscera
- Cyanosis (if respiratory failure preceded death)
- Signs of the route of exposure: skin excoriation (dermal), corrosive marks (if mixed with acid solvent - rare)
- Pulmonary oedema and congestion (frothy fluid in airways)
- Hyperaemic, congested mucosa of GI tract
- Petechial haemorrhages (hypoxia-related) - brain, epicardium, serosal surfaces
- Aspiration pneumonia in airways
- Liver congestion
- Brain oedema (in severe/fatal cases)
- Pulmonary oedema; bronchospasm evidence
- Evidence of aspiration (lipoid pneumonia - fat-laden macrophages)
- Neuronal degeneration (in OPIDN cases)
- Myocardial changes (ischaemia)
8D. Forensic Toxicology
| Sample | Analysis | Notes |
|---|---|---|
| Blood (antemortem or post-mortem) | RBC AChE activity; plasma BChE | Most important; must be frozen immediately |
| Urine | Metabolites (alkyl phosphates, p-nitrophenol for parathion) | Supports diagnosis; useful for specific compound ID |
| Gastric contents | Direct OP compound identification by GC-MS | Most important for homicide investigation |
| Vitreous humor | More stable than blood; less post-mortem redistribution | Useful when blood is unavailable |
| Liver, brain | Tissue OP levels | Post-mortem redistribution consideration |
| Hair | Chronic exposure | Longitudinal exposure history |
8E. Chemical Preservation of Evidence
- Gastric contents, blood, urine: collected in clean glass containers
- Sodium fluoride (NaF) added to blood samples - inhibits further AChE hydrolysis and preserves sample integrity
- All samples sealed, labelled, and maintained with proper chain of custody
- Refrigerate immediately (do not freeze blood for AChE if enzymatic assay needed)
8F. Nerve Agents (Chemical Warfare / Mass Casualty)
- Sarin (GB), Soman (GD), Tabun (GA), VX are OP nerve agents
- Used in chemical warfare and terrorism (Tokyo subway sarin attack 1995; Syrian conflict 2013)
- Extremely potent; death within minutes of inhalation or dermal exposure
- Soman "ages" within minutes - very narrow window for oxime therapy
- MARK 1 autoinjector (atropine + pralidoxime) issued to military personnel
- Pyridostigmine used as prophylaxis (reversible AChE inhibitor protecting active site from irreversible binding)
- Mass casualty management requires DECON stations, triage, rapid atropine and oxime deployment
8G. WHO Hazard Classification (Relevant to FMT)
| Class | Examples | Oral LD50 (rat) |
|---|---|---|
| Ia - Extremely hazardous | Parathion, TEPP | Very low |
| Ib - Highly hazardous | Methyl parathion, dichlorvos | Low |
| II - Moderately hazardous | Chlorpyrifos, coumaphos | Moderate |
| III - Slightly hazardous | Malathion, diazinon | High |
High WHO hazard class does not always predict severity in human cases due to variable exposure and metabolic differences.
8H. Medicolegal Documentation (FMT Checklist)
- Establish manner of death (suicide / accident / homicide)
- Record route of exposure and circumstances
- Document characteristic signs: miosis, excessive secretions, garlic odour
- Collect ante-mortem blood (NaF-preserved) and urine immediately
- At autopsy: collect gastric contents, peripheral blood, liver, brain, vitreous
- Identify the specific OP compound by GC-MS (toxicology)
- Confirm AChE inhibition (red cell AChE activity)
- Note proximity to pesticides at scene (suicide method, container)
- Exclude other causes of miosis (opiates, pontine lesion) in differential
- Maintain chain of custody for all samples
9. Differential Diagnosis of Cholinergic Toxidrome
| Feature | Organophosphate | Carbamate | Opioid | Pontine Haemorrhage |
|---|---|---|---|---|
| Miosis | Present | Present | Present | Present |
| Bradycardia | Present | Present | Present | Variable |
| Bronchospasm/secretions | Yes (prominent) | Yes | No | No |
| Fasciculations | Yes | Yes (milder) | No | No |
| Garlic odour | Yes | No | No | No |
| Naloxone response | No | No | Yes | No |
| Atropine response | Yes | Yes | No | No |
| AChE inhibition | Irreversible | Reversible (carbamylation) | No | No |
Carbamates cause reversible carbamylation of AChE (NOT phosphorylation); aging does NOT occur; pralidoxime is NOT necessary.
10. Summary: Key Points for PG FMT Paper
| Point | Fact |
|---|---|
| Mechanism | Irreversible phosphorylation of AChE → ACh accumulation → cholinergic crisis |
| Lethal dose | Variable by compound; parathion very toxic; malathion less so |
| Most common cause of death | Respiratory failure (bronchorrhea + respiratory muscle paralysis) |
| Diagnostic sign | Miosis (pinpoint pupils) + excessive secretions + garlic odour |
| Mnemonic for muscarinic effects | SLUDGE / DUMBELS / Killer Bs |
| Antidote 1 | Atropine (muscarinic antagonist; endpoint = dry secretions) |
| Antidote 2 | Pralidoxime 2-PAM (oxime; reactivates AChE; must give before aging) |
| Do NOT use | Succinylcholine (prolonged paralysis), beta-blockers |
| Intermediate syndrome | Day 1-5; respiratory paralysis; no cholinergic features; no response to atropine |
| OPIDN | 2-5 weeks; dying-back polyneuropathy; no treatment |
| Post-mortem sample | Gastric contents + peripheral blood (NaF) + urine + vitreous |
| Gold standard toxicology | GC-MS for compound ID; red cell AChE for inhibition |
| Most common manner of death | Suicide (India and South Asia - leading poisoning method) |
| Chemical warfare agents | Sarin, soman, tabun, VX; soman ages in minutes |
Tissue processing,fixation and staining , histopathjology long answe