Risk factors and pathophysiology of APLA
Reading File
Finding Sources
Searching PubMed
"antiphospholipid syndrome"[MeSH Terms] AND pathophysiology
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Searching the Web
antiphospholipid syndrome pathophysiology mechanisms 2024 2025 NETs complement beta2GPI
I now have comprehensive information from multiple authoritative sources. Let me compile the full, detailed answer.
Antiphospholipid Antibody Syndrome (APLA/APS): Risk Factors and Pathophysiology
Definition
Antiphospholipid syndrome (APS) is a systemic autoimmune thrombophilic disorder characterized by:
- Recurrent venous and/or arterial thrombosis
- Obstetric morbidity (recurrent pregnancy loss, fetal death, premature birth)
- Persistently positive antiphospholipid antibodies (aPL) on two or more occasions at least 12 weeks apart
The three canonical aPL antibodies are:
- Lupus anticoagulant (LA)
- Anticardiolipin antibodies (aCL) - IgG/IgM, moderate-to-high titer
- Anti-β2-glycoprotein I (anti-β2GPI) - IgG/IgM, moderate-to-high titer
"Triple positivity" (all three positive simultaneously) carries the highest thrombotic risk. - Creasy & Resnik's Maternal-Fetal Medicine
Risk Factors
1. Autoimmune Disease (Strongest Association)
- Systemic Lupus Erythematosus (SLE) is the most important predisposing condition. aPL antibodies are found in 25-75% of SLE patients; 34% are positive for lupus anticoagulant and 44% for anticardiolipin antibodies across published series. Most SLE patients with aPL never develop clinical APS - a "second hit" is required. - Brenner and Rector's The Kidney
- Other connective tissue diseases (rheumatoid arthritis, Sjögren's syndrome, systemic sclerosis) also confer risk.
2. Primary vs. Secondary APS
- Primary APS (30-55%): No associated autoimmune disease - aPL arise in isolation.
- Secondary APS: Occurs in the setting of SLE or other autoimmune conditions. - Brenner and Rector's The Kidney
3. Genetic/Immunogenetic Predisposition
- Among SLE patients, HLA-DRB1 loci are associated with susceptibility to developing aPL antibodies.
- Rare germline variants in complement regulatory genes (e.g., CFH, CFI) are found in ~60% of patients with catastrophic APS versus ~22% in thrombotic APS - highlighting a genetic complement dysregulation component. - Medscape/eMedicine; web search 2025
- Family members of APS patients have elevated risk of autoimmune disease and aPL positivity.
4. Infectious Triggers
- Infections (HIV, hepatitis C virus [HCV], syphilis, malaria, bacterial infections) can transiently induce aPL antibodies - but these infection-related antibodies are usually NOT associated with clinical APS (they lack β2GPI cofactor dependence).
- aPL are found in up to 2% of normal individuals and more frequently with infections. - Brenner and Rector's The Kidney
5. Drug-induced aPL
- Certain drugs (chlorpromazine, procainamide, phenytoin, hydralazine, quinidine) can induce anticardiolipin antibodies - again typically non-pathogenic, without clinical APS manifestations.
6. Hormonal / Physiologic "Second Hits"
These are factors that, in the presence of underlying aPL, tip the balance toward clinical thrombosis:
- Pregnancy (creates a hypercoagulable state: increased thrombin generation, impaired fibrinolysis via placental PAI-2, platelet activation)
- Oral contraceptive use and hormone replacement therapy
- Nephrotic syndrome (urinary loss of anticoagulant proteins S, C, antithrombin)
- Hyperlipidemia
- Immobilization, surgery, or trauma
- Intercurrent infection or inflammation - Brenner and Rector's The Kidney; Goldman-Cecil Medicine
7. Demographic Factors
- Female sex predominates (F:M ratio ~3-4:1), consistent with autoimmune disease predilection.
- Peak incidence in reproductive-age women.
- Non-white ethnicity may confer higher risk for worse outcomes in the context of SLE-associated APS.
Pathophysiology
The pathogenesis is multifactorial - aPL antibodies exert procoagulant effects at multiple simultaneous points. The "two-hit hypothesis" is central: aPL positivity alone is insufficient; a second trigger (infection, hormonal shift, complement activation) is needed to precipitate overt thrombosis.
Step 1: The Autoantigen - β2-Glycoprotein I (β2GPI)
The key autoantigen is β2GPI (also known as apolipoprotein H), a circulating plasma protein with 5 structural domains (DI-DV).
- Domain V (DV): Positively charged; binds to anionic phospholipids on activated endothelium, platelets, and apoptotic cells. This induces a conformational change from a closed circular to an open "J-shaped/fishhook" structure.
- Domain I (DI) epitope (Arg39-Arg43): Once β2GPI opens, this epitope is exposed and becomes the principal target of pathogenic autoantibodies. Anti-DI antibodies are specifically thrombogenic.
- Antibodies binding DI are highly specific for clinical APS; antibodies to other domains are less pathogenic.
Under normal conditions, phosphatidylserine (PS) is confined to the inner leaflet of the plasma membrane. When cells are activated or apoptotic, PS flips to the outer membrane surface - creating binding sites for β2GPI and, subsequently, aPL antibodies. - eMedicine/Medscape 2025
Step 2: Endothelial Cell Activation
Once aPL (particularly anti-β2GPI) binds to β2GPI on the endothelial surface:
- Upregulation of adhesion molecules: ICAM-1, VCAM-1, E-selectin are expressed on endothelium, promoting leukocyte-endothelial interactions and the inflammatory state.
- Tissue factor (TF) generation: Both monocytes and vascular endothelium generate TF upon aPL stimulation, directly initiating the extrinsic coagulation cascade.
- eNOS inhibition: aPL binding to DI of β2GPI impairs endothelial nitric oxide synthase (eNOS) phosphorylation, reducing NO synthesis. NO normally inhibits platelet aggregation and promotes vasodilation - its loss creates a prothrombotic, vasoconstrictive environment.
- MAPK and NF-κB pathway activation: aPL binding increases signaling through these pathways, amplifying pro-inflammatory cytokine production (TNF-α, IL-1β, IL-6). - Braunwald's Heart Disease; Metabolites 2025
Step 3: Platelet Activation
- aPL (especially anti-β2GPI) bind to β2GPI on the platelet surface, directly activating platelets.
- Activated platelets synthesize and release thromboxane A2 (TXA2), a potent platelet aggregator and vasoconstrictor - amplifying the procoagulant state.
- Platelets also expose anionic phospholipids on their surface, providing the "phospholipid scaffold" upon which coagulation complexes (tenase, prothrombinase) assemble more efficiently.
- The result is thrombocytopenia (platelet consumption in microthrombi) combined with paradoxical thrombosis. - Braunwald's Heart Disease
Step 4: Coagulation Cascade Interference
aPL antibodies interfere with multiple anticoagulant proteins:
| Target | Normal Function | Effect of aPL |
|---|---|---|
| Prothrombin | Coagulation factor | Autoantibody formation; stabilized on phospholipid membranes |
| Protein C | Anticoagulant (cleaves Va/VIIIa) | Inhibited, reducing anticoagulant activity |
| Protein S | Cofactor for Protein C | Inhibited |
| Annexin V | Forms anticoagulant shield on phospholipid surfaces | Displaced by aPL, exposing procoagulant phospholipids |
| Factor XII / Tissue-type plasminogen activator (tPA) | Fibrinolysis | Impaired fibrinolysis |
| mTORC pathway | Intracellular signaling | Inhibition → procoagulant effect |
- The net effect of coagulation protein interference is impaired fibrinolysis alongside enhanced clot formation.
- aPL binding to prothrombin can also inhibit thrombin inactivation, prolonging the procoagulant state.
- Lupus anticoagulant - paradoxically - prolongs the aPTT in vitro (competing for phospholipid in the assay) but causes thrombosis in vivo. - Brenner and Rector's The Kidney; Robbins Basic Pathology
Step 5: Complement Activation
This is a critical emerging mechanism:
- aPL (especially anti-β2GPI) activate the classical complement pathway, generating C5a and the membrane attack complex (MAC/C5b-9).
- C5a recruits and activates neutrophils and promotes endothelial injury.
- In pregnancy loss, complement activation at the placenta is a primary pathogenic mechanism - animal models show that inhibiting complement prevents fetal loss even in the presence of aPL.
- NETs (neutrophil extracellular traps): aPL-activated neutrophils undergo NETosis, releasing DNA-protein complexes that:
- Provide a scaffold for thrombus formation
- Act as a source of autoantigens (amplifying aPL production)
- Activate the complement cascade further
- Anti-NET antibodies in primary APS impair NET clearance, sustaining complement activation in a pathogenic positive-feedback loop.
- Catastrophic APS is strongly associated with complement regulatory gene variants (CFH, CFI), underscoring complement dysregulation as a driver of the most severe disease. - Braunwald's Heart Disease; Medscape 2025; web search (PMC 2025)
Step 6: The "Two-Hit Model" for Thrombosis
aPL positivity (Hit 1)
+
Second trigger (Hit 2): infection, pregnancy, OCP, complement activation, surgery
↓
Overt clinical thrombosis / obstetric event
This explains why:
- Most aPL-positive individuals never develop thrombosis
- Thrombotic events are often precipitated by identifiable triggers
- Infection-induced aPL (without β2GPI dependence) rarely cause APS
Obstetric Pathophysiology
The mechanisms of pregnancy loss differ somewhat from vascular thrombosis:
- Placental thrombosis - thrombosis of spiral arteries, intervillous space, and decidual vessels causing placental insufficiency and fetal death.
- Complement-mediated placental injury - aPL-induced C3a and C5a activation at the trophoblast-decidua interface causes:
- Direct trophoblast apoptosis and injury
- Reduced trophoblast invasiveness
- Impaired placentation and spiral artery remodeling
- Direct trophoblast dysfunction - aPL inhibit trophoblast differentiation and invasive capacity independently of thrombosis.
- Annexin V disruption - displaces the anticoagulant annexin V shield on trophoblast surfaces, promoting local clotting. - Creasy & Resnik's Maternal-Fetal Medicine
Histopathology
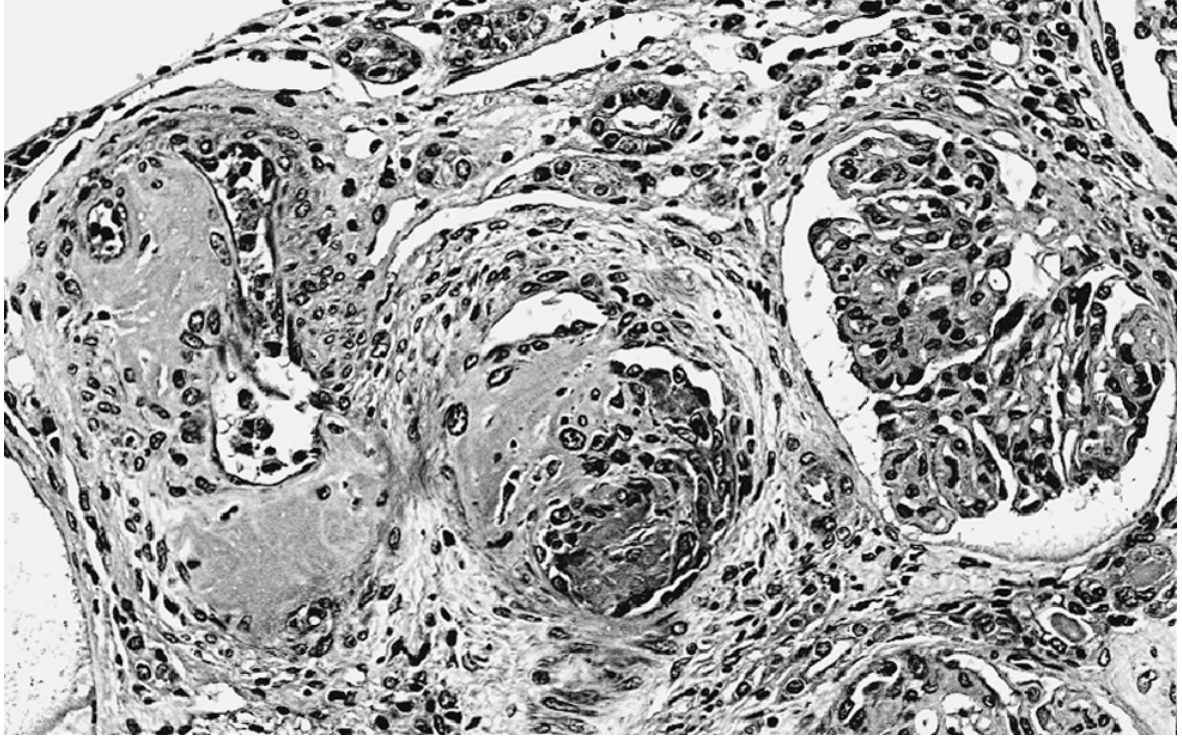
APL nephropathy: Organizing recanalized thrombi narrowing the lumens of interlobular arteries. The adjacent glomerulus shows ischemic-type retraction of its tuft. Arteries and arterioles show thrombosis PLUS reactive intimal mucoid thickening, subendothelial fibrosis, and medial hyperplasia (HE, x200). - Brenner and Rector's The Kidney
Summary Diagram of Pathophysiology
Susceptible host (HLA-DRB1, complement gene variants)
↓
Exposure: infection, drugs, autoimmune trigger
↓
aPL production (anti-β2GPI, LA, aCL)
↓
β2GPI binds anionic phospholipids → opens to "J" shape
↓
aPL binds Domain I of β2GPI on endothelium / platelets / monocytes
↓
┌──────────────────────────────────────┐
│ Endothelial activation: │
│ ↑TF, ↑adhesion molecules, ↓eNOS │
│ │
│ Platelet activation: │
│ ↑TXA2, ↑phospholipid scaffold │
│ │
│ Coagulation protein disruption: │
│ ↓Protein C/S, ↓Annexin V, ↓tPA │
│ │
│ Complement activation: │
│ C5a → neutrophil activation → NETs │
└──────────────────────────────────────┘
↓
Second hit (pregnancy, infection, OCP...)
↓
THROMBOSIS / PREGNANCY LOSS
Recent Evidence Update (2024-2025)
| PMID | Title | Key Update |
|---|---|---|
| 38702511 | "Rethinking antiphospholipid syndrome" (Nat Rev Rheumatol 2024) | Proposes reclassification based on pathogenic mechanism rather than antibody type alone |
| 40418947 | "Towards precision medicine in APS" (Lancet Rheumatol 2025) | Identifies molecular endotypes for targeted therapy (complement inhibitors, BTK inhibitors) |
| 40386969 | "APS: Emerging Therapeutic Opportunities" (Arthritis Rheumatol 2025) | Highlights NETs, complement, and B-cell targeting as novel therapeutic directions |
Note: These recent reviews emphasize that complement inhibition (e.g., eculizumab) and NET-targeting strategies are emerging as therapeutic options, particularly in catastrophic APS - suggesting these pathways are not only mechanistically central but also clinically actionable.
Sources: Brenner and Rector's The Kidney; Braunwald's Heart Disease; Goldman-Cecil Medicine; Creasy & Resnik's Maternal-Fetal Medicine; Robbins Basic Pathology; Metabolites 2025; Nat Rev Rheumatol 2024; Lancet Rheumatol 2025.
This is a shared conversation. Sign in to Orris to start your own chat.