Parkinsons disease
Reading File
Finding Sources
Searching PubMed
"Parkinson disease"[MeSH Terms] AND management
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive material. Let me compile the full, authoritative response on Parkinson's Disease.
Parkinson's Disease
Overview
Parkinson's disease (PD) is a progressive neurodegenerative disorder and the second most common neurodegenerative disease after Alzheimer's. Symptoms typically emerge between 55 and 65 years of age. It was first formally described by James Parkinson in 1817 as the "shaking palsy." - Bradley and Daroff's Neurology in Clinical Practice
Pathophysiology
The hallmark pathology involves degeneration of pigmented dopaminergic neurons in the substantia nigra pars compacta. By the time motor symptoms appear, approximately 60-80% of these neurons have already been lost.
The Dopamine-Acetylcholine Imbalance
Normally, the neostriatum maintains a balance between:
- Inhibitory dopaminergic neurons (from substantia nigra)
- Excitatory cholinergic neurons (within neostriatum)
In PD, destruction of substantia nigra cells depletes dopamine in the neostriatum. The loss of dopamine's inhibitory influence allows cholinergic neurons to become relatively overactive, triggering a chain of abnormal signaling leading to motor dysfunction.
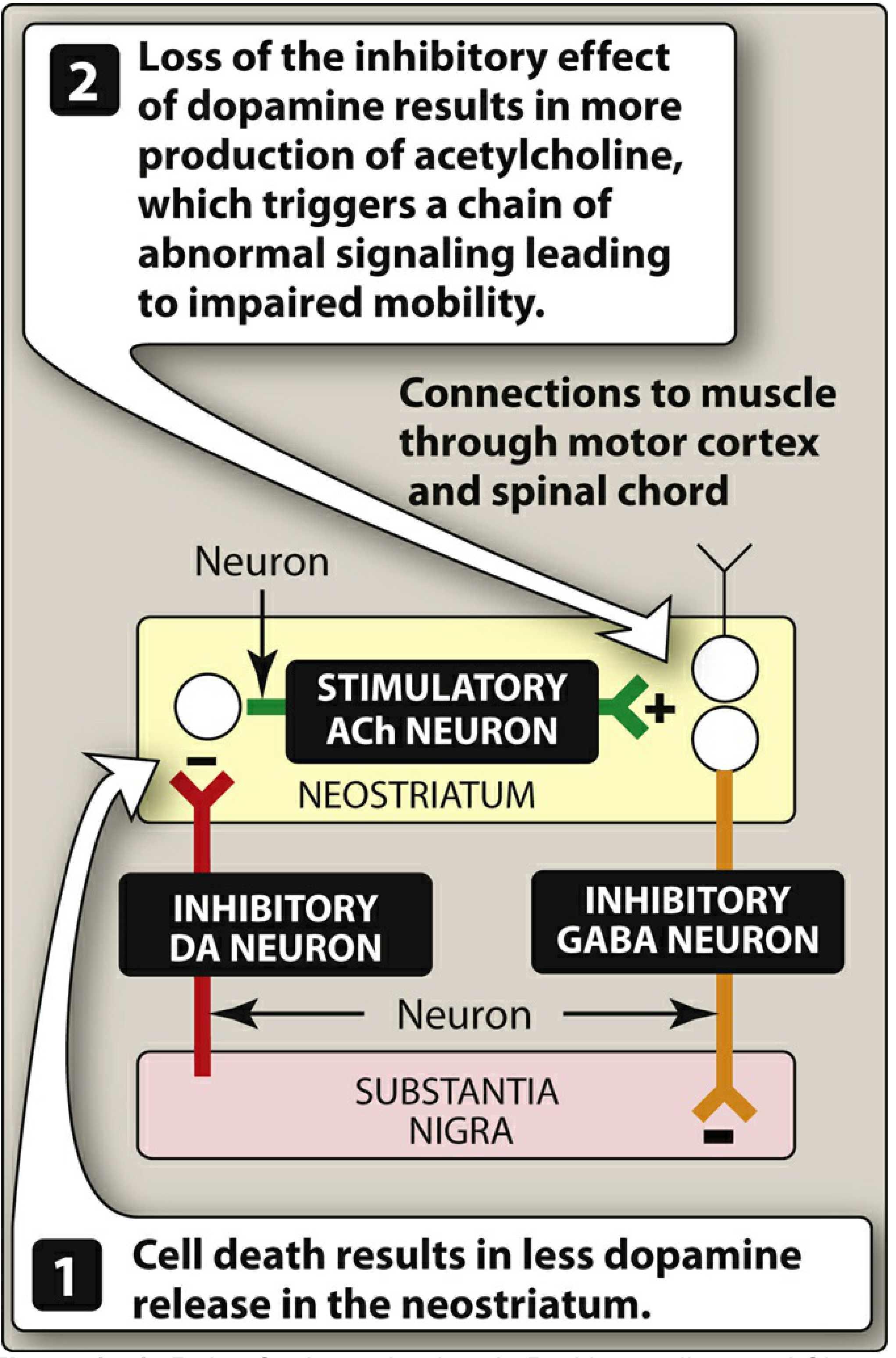
Figure: Role of the substantia nigra in PD. Cell death causes less dopamine release in the neostriatum, leading to unchecked ACh activity and impaired mobility. - Lippincott Illustrated Reviews: Pharmacology
Basal Ganglia Circuit Disruption
Dopamine acts on two opposing pathways in the striatum:
- Direct pathway (D1 receptors): dopamine is excitatory - facilitates movement
- Indirect pathway (D2 receptors): dopamine is inhibitory - reduces the brake on movement
In PD, dopamine loss reduces excitation via the direct pathway AND reduces inhibition via the indirect pathway - both effects converge to produce excessive inhibition of the thalamus, resulting in the hypokinetic features of PD. - Costanzo Physiology 7e
Alpha-Synuclein and Lewy Bodies
The pathological hallmark on histology is the Lewy body - an intracytoplasmic inclusion with a dense eosinophilic core and a lighter halo, found within surviving dopaminergic neurons. Lewy bodies and Lewy neurites result from abnormal aggregation of alpha-synuclein protein. They may also contain neurofilaments, parkin, and ubiquitin.
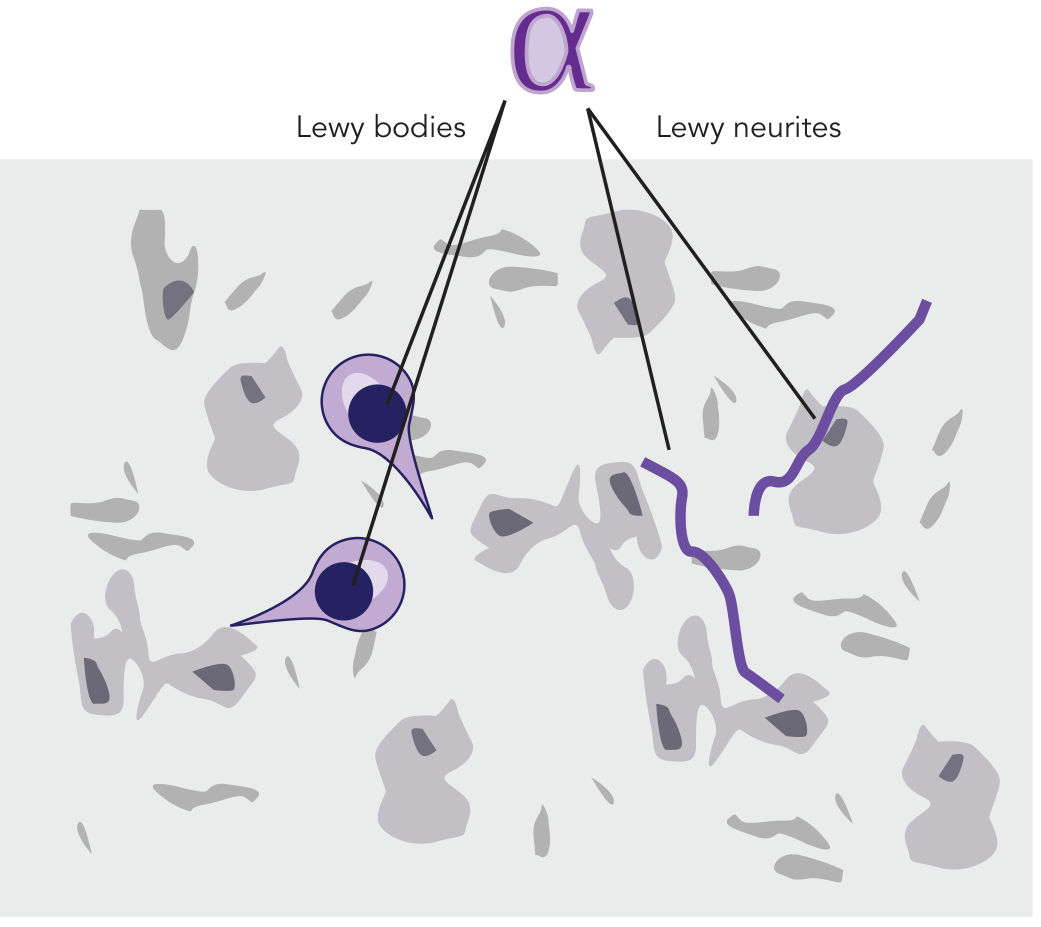
Figure: Lewy bodies (intracellular) and Lewy neurites (in neuronal processes) - both composed of aggregated alpha-synuclein. - Stahl's Essential Psychopharmacology
Neuronal loss is not confined to the substantia nigra - it also occurs in the locus coeruleus, raphe nuclei, dorsal motor nucleus of the vagus, olfactory bulb, and enteric nervous system.
Clinical Features
Motor Symptoms (the classic tetrad)
| Feature | Description |
|---|---|
| Resting tremor | "Pill-rolling" tremor at 4-6 Hz, suppressed by voluntary movement |
| Rigidity | Increased tone throughout range of motion; often "cogwheel" type |
| Bradykinesia | Slowness of voluntary movement; most disabling feature |
| Postural instability | Impaired reflex adjustments; late feature, leads to falls |
Other Motor Manifestations
- Masked facies (hypomimia) - reduced facial expression, decreased blink rate
- Micrographia - progressively smaller handwriting
- Hypophonia - soft, monotone voice with hurried muttering quality
- Festinating gait - small, shuffling steps; difficulty initiating (freezing), tendency to lean forward (anteropulsion) or backward (retropulsion)
- En bloc turning - turning without normal torso twist
- Myerson's sign - inability to suppress blinking when glabella is repeatedly tapped (non-specific)
- Dysphagia - recognized by Parkinson himself in 1817; subjective prevalence ~35%, objective up to 82%; involves oral, pharyngeal, and esophageal phases; silent aspiration in 15-33%
Non-Motor Symptoms (often precede motor symptoms)
- Autonomic: orthostatic hypotension, constipation, urinary urgency, sexual dysfunction, seborrhea
- Sleep: REM sleep behavior disorder (RBD) - often an early prodromal marker
- Olfactory: hyposmia/anosmia (may predate motor symptoms by years)
- Neuropsychiatric: depression (most common psychiatric disturbance), anxiety, apathy, psychosis (typically medication-induced visual hallucinations in ~40%, delusions in ~16%)
- Cognitive: mild executive dysfunction is common early; dementia occurs in 15-40% later in the course
Note on psychosis: In PD, hallucinations are usually fleeting and nocturnal, motor symptoms virtually always precede psychosis, and psychosis is generally medication-induced. This distinguishes it from Dementia with Lewy Bodies (DLB), where psychosis is a core feature occurring even without medications. - Bradley and Daroff's Neurology in Clinical Practice
Diagnosis
PD is a clinical diagnosis based on the presence of bradykinesia plus at least one of: resting tremor or rigidity. There is no definitive antemortem biological test.
Supporting features:
- Asymmetric onset
- Good response to levodopa
- Progressive course over 5-15 years
Investigations:
- DaT SPECT (dopamine transporter scan): shows reduced uptake in the striatum; useful when diagnosis is uncertain
- MRI brain: typically normal in idiopathic PD; used to exclude structural causes
Red flags suggesting alternative diagnoses:
- Early postural instability (before 3 years)
- Rapid progression
- Symmetrical onset
- No response to levodopa
- Early falls, early dementia, early autonomic failure, supranuclear gaze palsy, cerebellar signs
Up to 30% of PD patients never develop tremor, so its absence does not exclude the diagnosis. - Neuroanatomy Through Clinical Cases, 3e
Pharmacological Treatment
Drug therapy aims to restore the dopamine/acetylcholine balance in the basal ganglia. Currently available drugs offer only symptomatic relief - none have proven neuroprotective or disease-modifying effects.
1. Levodopa + Carbidopa (first-line, most effective)
Levodopa is the metabolic precursor of dopamine. It crosses the blood-brain barrier and is converted to dopamine by surviving substantia nigra neurons.
Carbidopa is a peripheral dopa-decarboxylase inhibitor that:
- Reduces peripheral conversion of levodopa to dopamine (preventing nausea, vomiting, cardiac arrhythmias)
- Increases CNS availability of levodopa
- Allows a 75% reduction in levodopa dose
Problems with long-term levodopa use:
- Wearing-off phenomenon: as neuronal loss progresses, the drug effect duration shortens between doses
- On-off fluctuations: unpredictable swings between mobile ("on") and immobile ("off") states
- Dyskinesias: involuntary choreiform movements, typically at peak dose
- Nausea, orthostatic hypotension, psychosis
2. COMT Inhibitors (adjunct to levodopa)
Entacapone, opicapone, tolcapone inhibit catechol-O-methyltransferase, which normally converts levodopa to 3-O-methyldopa (a metabolite that competes with levodopa for CNS transport). By blocking this pathway, COMT inhibitors:
- Increase central levodopa uptake
- Raise brain dopamine levels
- Reduce "wearing-off" effects
Tolcapone carries a risk of fulminant hepatic necrosis and is reserved for cases where other agents have failed. Entacapone and opicapone are preferred and lack this hepatotoxicity. - Lippincott Illustrated Reviews: Pharmacology
3. MAO-B Inhibitors
Selegiline and rasagiline selectively inhibit MAO type B, which is the primary enzyme for dopamine catabolism in the brain. This prolongs dopamine availability. They may be used as initial monotherapy in mild disease or as adjuncts to levodopa.
4. Dopamine Receptor Agonists
Non-ergot agents (preferred): ropinirole, pramipexole, rotigotine, apomorphine
Ergot derivative (less used): bromocriptine
These agents directly stimulate dopamine receptors. Advantages over levodopa:
- Longer duration of action
- Lower risk of dyskinesia
- Useful in patients with levodopa fluctuations
- Initial monotherapy in younger patients delays dyskinesia
Adverse effects: nausea, orthostatic hypotension, somnolence, hallucinations, impulse control disorders (gambling, hypersexuality - particularly with pramipexole/ropinirole).
5. Anticholinergic Agents
Benztropine, trihexyphenidyl - reduce cholinergic overactivity. Primarily useful for tremor in younger patients. Cause significant anticholinergic side effects (confusion, memory impairment, urinary retention) and are generally avoided in the elderly.
6. Amantadine
An NMDA receptor antagonist with mild dopaminergic and anticholinergic properties. Used for mild early symptoms and, importantly, for reducing levodopa-induced dyskinesias in advanced disease.
Non-Pharmacological and Surgical Treatment
Exercise and Physiotherapy
A 2025 network meta-analysis (PMID: 39880702) found that specific exercise types and doses improve motor symptoms in PD. Resistance training, treadmill training, and dance therapy have shown benefits for gait, balance, and bradykinesia.
Deep Brain Stimulation (DBS)
The most effective surgical option. Electrodes are implanted in the subthalamic nucleus (STN) or globus pallidus interna (GPi). DBS is indicated for:
- Advanced PD with motor fluctuations refractory to medical therapy
- Intolerable levodopa-induced dyskinesias
- Preserved cognitive function
DBS continuously modulates the cortico-striato-thalamo-cortical circuit, reducing pathological beta oscillations and restoring more physiological motor cortex excitability. The effect on tremor, rigidity, and bradykinesia may accumulate over hours of stimulation. - Bradley and Daroff's Neurology in Clinical Practice
Repetitive Transcranial Magnetic Stimulation (rTMS)
Investigational. Rationale is to increase motor cortex excitability and suppress pathological beta oscillations. Results of class 1 trials remain ambiguous. Larger RCTs are needed.
Swallowing and Speech Therapy
Given that dysphagia affects up to 82% objectively and silent aspiration occurs in 15-33%, speech and language therapy is an important component. Aspiration pneumonia is a leading cause of death in advanced PD. REM sleep behavior disorder management and fall prevention are also key non-pharmacological targets.
Complications and Prognosis
- Disease progression is typically insidious, over 5-15 years
- Many patients die of other causes (given late onset) rather than directly from PD
- Leading causes of death in advanced PD: aspiration pneumonia, falls/trauma, immobility complications
- Dementia (PD dementia) occurs in 15-40% of patients late in the course
- PD dementia shares pathological overlap with DLB (both are "synucleinopathies")
Key Differential Diagnoses
| Condition | Distinguishing Features |
|---|---|
| Drug-induced parkinsonism | History of dopamine blockers (antipsychotics, metoclopramide); usually reversible |
| Multiple System Atrophy (MSA) | Early autonomic failure, cerebellar signs, poor levodopa response |
| Progressive Supranuclear Palsy (PSP) | Early postural instability, supranuclear gaze palsy, axial rigidity |
| Corticobasal Degeneration (CBD) | Alien limb, asymmetric apraxia, cortical sensory loss |
| Dementia with Lewy Bodies (DLB) | Dementia precedes or co-occurs with parkinsonism, fluctuating cognition, spontaneous psychosis |
| Essential Tremor | Action/postural tremor, no bradykinesia or rigidity, family history |
Recent Evidence (2024-2026)
- Exercise for motor symptoms (PMID: 39880702, 2025 network meta-analysis): Specific exercise modalities significantly improve motor outcomes - exercise should be considered a core therapeutic component, not optional.
- Virtual reality for balance (PMID: 40142335, 2025 meta-analysis): VR combined with conventional therapy improves balance compared to conventional therapy alone.
- Anxiety management (PMID: 40568748, 2025): Anxiety affects up to 50% of PD patients and remains undertreated; a recent meta-analysis reviewed pharmacological and non-pharmacological approaches.
Sources: Bradley and Daroff's Neurology in Clinical Practice | Lippincott Illustrated Reviews: Pharmacology | Neuroanatomy Through Clinical Cases 3e | Stahl's Essential Psychopharmacology | Costanzo Physiology 7e | Sleisenger and Fordtran's GI Disease
Pls provide me Harrison based for sure
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
I now have all the content I need from Harrison's 22e. Let me compile the comprehensive Harrison-based answer.
Parkinson's Disease
Harrison's Principles of Internal Medicine, 22nd Edition (2025)
Authors: C. Warren Olanow, Anthony H.V. Schapira, Christine Klein
Epidemiology
PD is the second most common age-related neurodegenerative disease, exceeded only by Alzheimer's disease. Key epidemiological facts from Harrison's:
- Estimated ~10.8 million people with PD worldwide; expected to double within 20 years due to population aging
- Mean age of onset: ~60 years (but can occur in the 20s, especially with pathogenic gene mutations)
- Lifetime risk: ~3% for men, ~2% for women
- Frequency increases steeply with age
Pathology
The hallmark pathological features of PD are:
- Degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc)
- Reduced striatal dopamine
- Lewy pathology - intraneuronal proteinaceous inclusions (Lewy bodies in cell bodies; Lewy neurites in axons) that stain positive for alpha-synuclein (Fig. 446-1)
Neurodegeneration is not limited to the SNc. Lewy pathology also affects:
- Cholinergic neurons of the nucleus basalis of Meynert (NBM)
- Norepinephrine neurons of the locus coeruleus (LC)
- Serotonin neurons of raphe nuclei
- Neurons of the olfactory system, cerebral hemispheres, spinal cord, and peripheral autonomic nervous system
This widespread pathology explains the many non-motor features of PD.
Premotor Stage ("Body-First vs. Brain-First" PD)
Harrison's 22e highlights a key recent concept: clinical symptoms reflecting early nondopaminergic involvement - constipation, anosmia, REM sleep behavior disorder (RBD), and cardiac denervation - can precede the onset of classic motor features by several years to decades. These are now considered to represent an early premotor form of the disease, not merely risk factors. Two new biological classification systems have been developed:
- Based on neuronal alpha-synuclein accumulation staging
- Based on alpha-synuclein deposition + distribution of neurodegeneration + pathogenic gene variants
Etiology and Pathogenesis
Genetics
| Gene | Inheritance | Notes |
|---|---|---|
| SNCA (alpha-synuclein) | Autosomal dominant | Point mutations and duplications/triplications; multiplications increase expression and severity |
| LRRK2 (leucine-rich repeat kinase 2) | Autosomal dominant | p.G2019S is the most common mutation; founder effect in Ashkenazi Jewish and North African Berber populations |
| PRKN (Parkin) | Autosomal recessive | Most common AR cause; early-onset PD (median AAO ~32 years) |
| PINK1 | Autosomal recessive | Mitochondrial kinase; early-onset |
| PARK7 (DJ-1) | Autosomal recessive | Rare; early-onset |
| GBA1 (glucocerebrosidase) | Risk factor | Heterozygous mutations are the most common genetic risk factor for PD |
Alpha-synuclein prion hypothesis: Based on experimental evidence (injection of PD brain homogenates triggers spread of pathology in mice; Lewy pathology develops in fetal nigral cell grafts transplanted into PD patients), Harrison's describes the hypothesis that alpha-synuclein may behave like a prion - misfolded protein that self-propagates and spreads from cell to cell in a stereotyped pattern (Braak staging).
Putative Mechanisms of Cell Death
- Oxidative stress (increased free radicals from dopamine metabolism)
- Mitochondrial dysfunction (Complex I deficiency in SNc)
- Proteasomal/lysosomal dysfunction - failure to clear alpha-synuclein aggregates
- Neuroinflammation - activated microglia in SNc
- Apoptosis as a final common pathway
MPTP Model
A key insight came from drug abusers who developed acute parkinsonism after exposure to MPTP (a contaminant of synthetic heroin). MPTP is converted by MAO-B to MPP+, which selectively damages dopamine neurons by inhibiting mitochondrial Complex I. This model validates the role of mitochondrial dysfunction and provided the rationale for MAO-B inhibitors as potential neuroprotective agents.
Clinical Features
Cardinal Motor Features (TABLE 446-1)
| Cardinal Features | Other Motor Features | Nonmotor Features |
|---|---|---|
| Bradykinesia | Micrographia | Anosmia |
| Rest tremor | Masked facies (hypomimia) | Sensory disturbances (pain, hyposmia) |
| Rigidity | Reduced eye blinking | Mood disorders (depression, anxiety, apathy) |
| Postural instability | Drooling | Sleep disturbances (fragmented sleep, RBD) |
| Hypophonia | Autonomic disturbances | |
| Dysphagia | Orthostatic hypotension | |
| Freezing of gait | GI disturbances | |
| Falling | Genitourinary disturbances | |
| Cognitive impairment/dementia | ||
| Psychosis (hallucinations, delusions) |
Diagnosis - UK Brain Bank Criteria & MDS Criteria
Historically, PD was diagnosed based on 2 of 3 parkinsonian features (tremor, rigidity, bradykinesia). However, postmortem studies found a 24% error rate with these criteria alone.
The UK Brain Bank Criteria - combining bradykinesia + rigidity + rest tremor + asymmetry + good levodopa response - improved pathological confirmation to >90% of cases.
The newer MDS Clinical Diagnostic Criteria (Movement Disorder Society) retain motor parkinsonism as the core feature and add:
- Supportive criteria (features increasing diagnostic confidence)
- Absolute exclusion criteria
- Red flags (must be counterbalanced by supportive criteria)
Two diagnostic certainty levels: Clinically Established PD and Clinically Probable PD.
Imaging
DaT PET/SPECT: Shows reduced and asymmetric uptake in the striatum, particularly in the posterior putamen with relative sparing of the caudate nucleus (Fig. 446-3). This reflects degeneration of dopaminergic nerve terminals projecting from the SNc.
In atypical parkinsonism (MSA, PSP): striatal dopamine depletion can also be seen, so DaT imaging does NOT reliably distinguish PD from atypical parkinsonism. However, FDG-PET of basal ganglia/thalamic network may help - PD shows decreased GPI activity with increased thalamic activity (opposite in atypical parkinsonism).
Differential Diagnosis
Atypical Parkinsonism (TABLE 446-3 from Harrison's)
Features suggesting atypical or secondary parkinsonism:
| Symptom/Sign | Alternative Diagnosis |
|---|---|
| Early speech and gait impairment, no tremor, no asymmetry, early falls | Atypical parkinsonism (MSA, PSP) |
| Neuroleptic/metoclopramide exposure | Drug-induced parkinsonism |
| Onset before age 40 | Genetic PD, Wilson's disease, DRD |
| Liver disease | Wilson's disease |
| Dementia/hallucinations preceding motor features | Dementia with Lewy Bodies (DLB) |
| Diplopia, impaired vertical (downward) gaze | PSP |
| Poor or no response to adequate levodopa trial | Atypical or secondary parkinsonism |
MSA (Multiple System Atrophy): Parkinsonism + cerebellar signs + prominent autonomic dysfunction; alpha-synuclein accumulates in oligodendrocytes (GCIs), not neurons; MRI shows putaminal rim, hot cross bun sign.
PSP (Progressive Supranuclear Palsy): Parkinsonism + slow saccades + restricted downward gaze + early falls (hyperextension of neck); tau pathology (neurofibrillary tangles); hummingbird sign on MRI.
Drug-induced parkinsonism: Harrison's specifically flags metoclopramide - physicians must be aware that drugs used for GI problems are neuroleptics and can cause secondary parkinsonism.
Dopa-Responsive Dystonia (DRD): Especially in those <20 years with parkinsonism; GTP-cyclohydrolase 1 mutation; responds to levodopa but no degeneration on FD-PET and no drug-induced dyskinesias.
Wilson's disease must always be ruled out in young-onset parkinsonism, as progression can be prevented with copper chelators.
Treatment
LEVODOPA - The Gold Standard
"Levodopa remains the most effective symptomatic treatment for PD and the gold standard against which new therapies are compared." - Harrison's 22e
Historical background: Carlsson (late 1950s) showed reserpine-induced parkinsonism in rabbits was reversed by levodopa. Hornykiewicz demonstrated striatal dopamine deficiency in PD patients, suggesting dopamine replacement therapy.
Mechanism: Levodopa crosses the BBB (dopamine cannot); it is converted to dopamine in surviving SNc neurons, restoring striatal dopamine.
Peripheral decarboxylase inhibitors are always co-administered:
- Carbidopa (USA - Sinemet)
- Benserazide (most other countries - Madopar)
- Prevent peripheral levodopa conversion to dopamine, reducing: nausea, vomiting, orthostatic hypotension (mediated via area postrema dopamine receptors outside the BBB)
Available formulations (Harrison's 22e - updated list):
- Standard oral levodopa/carbidopa (Sinemet)
- Controlled-release (Sinemet CR / Madopar HP)
- Long-acting oral formulation (Rytary)
- Levodopa/carbidopa/entacapone combined (Stalevo)
- Levodopa-carbidopa intestinal gel - continuous intra-intestinal infusion
- Continuous subcutaneous infusions of levodopa
- Inhaled levodopa - absorbed through pulmonary alveoli (approved on-demand therapy for off periods)
Motor Complications of Levodopa (Fig. 446-6)
With ongoing disease progression and chronic levodopa use, most patients develop motor complications:
-
Wearing-off phenomenon: Duration of benefit per dose progressively shortens. Initially, benefit lasts many hours (the "long-duration response"). Eventually, benefit approaches the drug's half-life (60-90 min).
-
On-off phenomenon: Rapid, unpredictable switches between the "on" state (drug working) and "off" state (parkinsonism returns).
-
Dyskinesias: Involuntary choreiform/dystonic movements, typically at peak dose during "on" periods.
In advanced cases, the response to an individual dose may be variable and unpredictable - full-on, partial-on, delayed, or even dose failure.
On-demand therapies for off periods (3 FDA-approved):
- Inhaled levodopa (Inbrija)
- Subcutaneous apomorphine injection (Apokyn)
- Sublingual apomorphine (Kynmobi)
All are fast-acting, avoid variable oral bioavailability, and provide predictable return to the on state.
Other Pharmacological Agents
| Drug Class | Examples | Role |
|---|---|---|
| MAO-B inhibitors | Selegiline, rasagiline | Early monotherapy or adjunct; potential neuroprotective effect (ADAGIO trial - rasagiline 1 mg/d showed disease-modifying consistent results) |
| Dopamine agonists | Pramipexole, ropinirole, rotigotine, apomorphine | Monotherapy in early disease (especially younger patients); adjunct in fluctuating disease; longer duration than levodopa; lower dyskinesia risk |
| COMT inhibitors | Entacapone, opicapone, tolcapone | Reduce wearing-off; tolcapone reserved for refractory cases (hepatotoxicity risk) |
| Amantadine | NMDA antagonist | Mild early PD; importantly, reduces levodopa-induced dyskinesias |
| Anticholinergics | Benztropine, trihexyphenidyl | Primarily for tremor in younger patients; avoid in elderly |
Neuroprotection - The Major Unmet Need
"A neuroprotective or disease-modifying therapy that slows or stops disease progression remains the major unmet therapeutic need." - Harrison's 22e
Current investigational targets:
- Agents preventing toxic alpha-synuclein species formation or accumulation
- LRRK2 inhibitors
- GCase (GBA1) enhancers
- GLP-1 agonists (developed for diabetes; anti-inflammatory and pro-mitochondrial effects; results in double-blind trials have been inconsistent)
The DATATOP study showed selegiline delayed disability requiring levodopa introduction, but could not definitively prove neuroprotection vs. symptomatic masking. The ADAGIO study using a delayed-start design showed rasagiline 1 mg/d had benefits consistent with a disease-modifying effect, but the 2 mg dose failed - reason remains uncertain.
Surgical Treatment
History: Early surgery involved motor cortex lesions (improved tremor but caused motor deficits - abandoned). Then VIM thalamotomy was found to reduce contralateral tremor without motor deficits, but had no effect on bradykinesia or rigidity.
Deep Brain Stimulation (DBS) - current standard surgical approach:
- Primary targets: subthalamic nucleus (STN) or globus pallidus interna (GPi)
- STN-DBS: reduces all cardinal motor features; often allows levodopa dose reduction
- GPi-DBS: particularly effective for dyskinesias
- Indications: Advanced PD with motor fluctuations refractory to optimized medical therapy; intolerable dyskinesias; preserved cognitive function
- Contraindications/poor candidates: Significant dementia, prominent non-dopaminergic features (freezing, falls), active psychiatric illness, unrealistic expectations
- Focused ultrasound thalamotomy (non-invasive): now available as an alternative for tremor-predominant PD
Emerging / Experimental Therapies (Harrison's 22e)
Cell-based/Stem Cell Therapies: Two NIH-sponsored trials of fetal midbrain cell grafts in the 1990s failed - only a small subset of younger patients showed benefit, and some developed graft-induced dyskinesias (attributed to serotonergic neurons inadvertently included in grafts). A newer European trial was terminated due to slow enrollment and insufficient cell harvest. Current excitement centers on iPSC-derived dopamine neurons and embryonic stem cell-derived neurons:
- Japan (Kyoto University): Phase 1/2 trial since 2018; 7 patients treated
- USA: FDA approved first clinical trial of ESC-derived dopamine neurons; 10 patients in Phase 1
- Europe: Trial of ESC-derived neurons began 2023; 4 patients on low dose with no acute adverse effects
Non-Motor Management
Harrison's emphasizes that nondopaminergic features (especially falls and dementia) are the primary source of disability and main reason for hospitalization and nursing home placement in the levodopa era - not motor symptoms.
Key non-motor domains:
- Depression: Most common psychiatric disturbance; SSRIs are first-line (though may worsen motor symptoms); tricyclics as alternative
- Psychosis: Visual hallucinations in up to 40% - usually medication-induced; clozapine (low dose) or pimavanserin (5-HT2A inverse agonist, does not worsen parkinsonism) are options
- Dementia: 15-40% develop dementia; rivastigmine (ChEI) has some evidence
- Autonomic: Orthostatic hypotension (fludrocortisone, midodrine, droxidopa); constipation; urinary dysfunction
- Sleep/RBD: Melatonin or low-dose clonazepam for RBD
- Anosmia: No treatment; important as a premotor marker
Source: Harrison's Principles of Internal Medicine, 22nd Edition (2025) - Chapter 446: Parkinson's Disease and Related Disorders. Authors: C. Warren Olanow, Anthony H.V. Schapira, Christine Klein.
This is not much to write in pg exam
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Here is the full PG exam-format answer, entirely from Harrison's 22e (Chapter 446):
PARKINSON'S DISEASE
Harrison's Principles of Internal Medicine, 22nd Edition — Chapter 446
C. Warren Olanow | Anthony H.V. Schapira | Christine Klein
1. DEFINITION & EPIDEMIOLOGY
- 2nd most common age-related neurodegenerative disease (after Alzheimer's)
- First described by James Parkinson in 1817 as "shaking palsy"
- Prevalence: ~10.8 million worldwide; expected to double in 20 years
- Mean age of onset: ~60 years; can occur in 20s with gene mutations
- Lifetime risk: 3% men, 2% women
- ~15% of cases are familial; the rest are sporadic
2. PATHOLOGY (FIGURE 446-1 - Harrison's)
Gross pathology
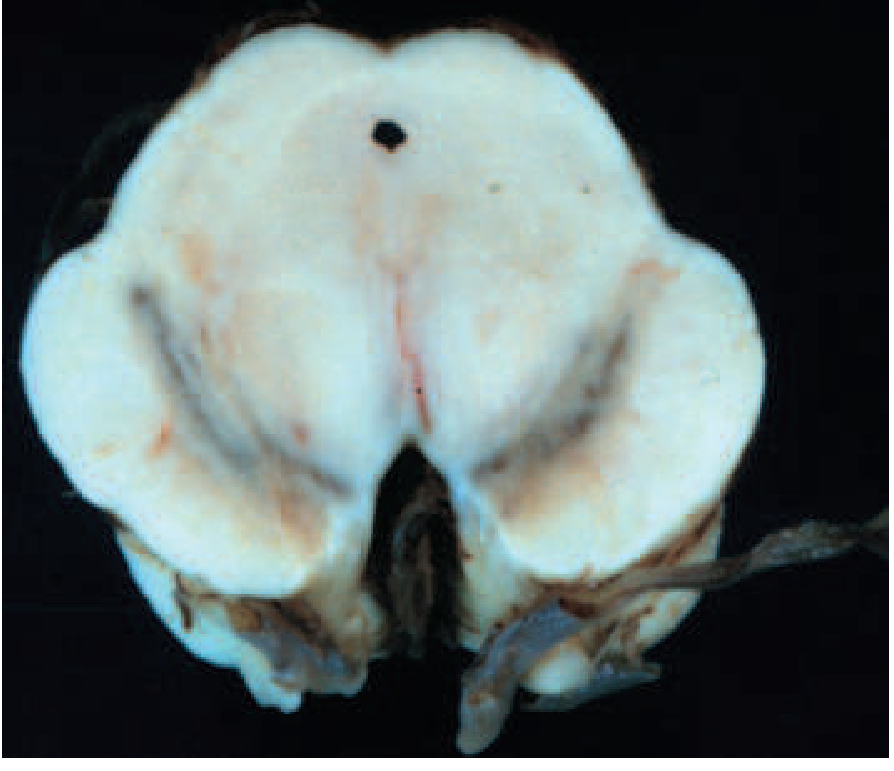
FIGURE 446-1A: Cross-section of PD midbrain (right) vs. normal (left) - note the striking pallor/depigmentation of the substantia nigra pars compacta due to loss of melanized dopaminergic neurons.
Microscopic pathology
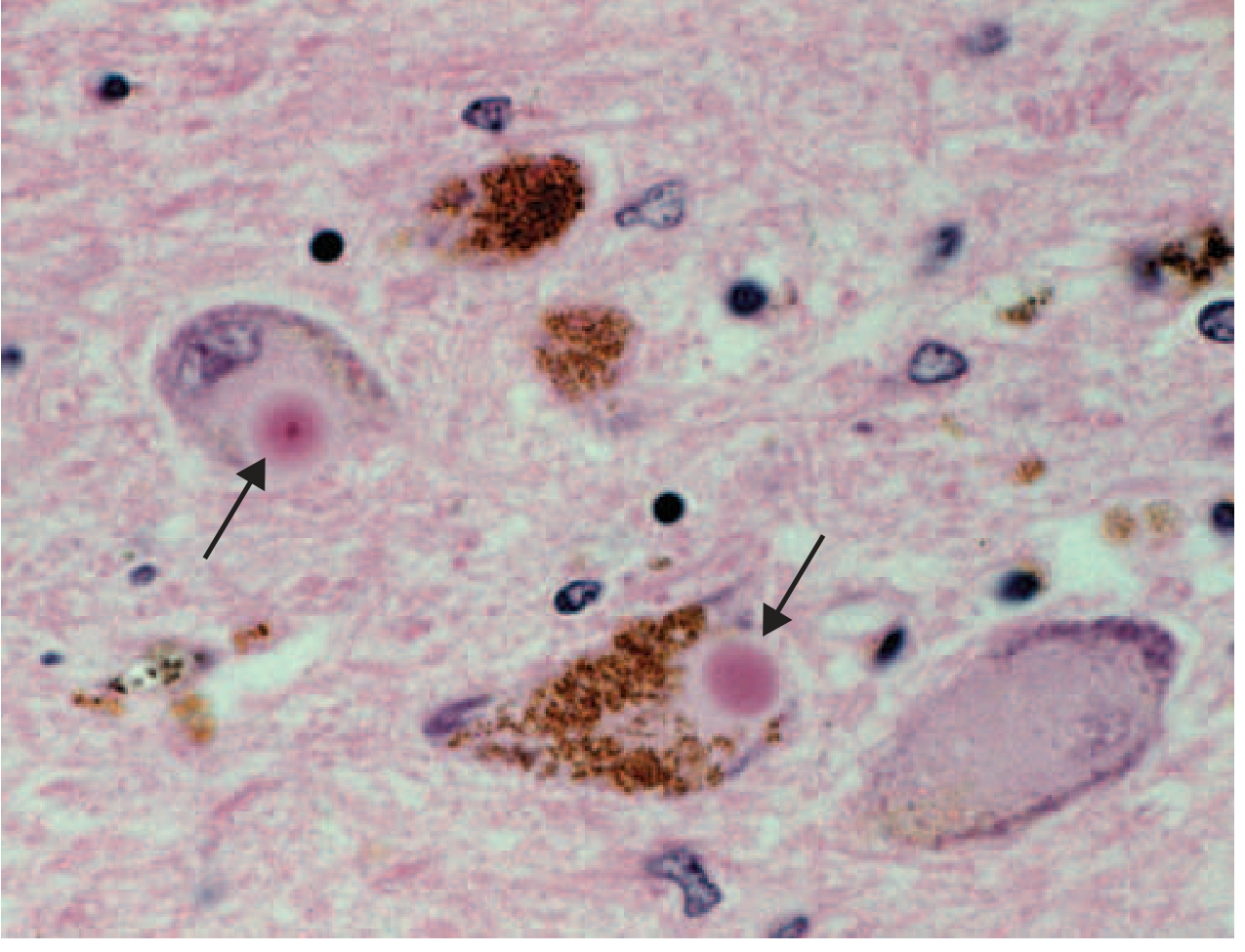
FIGURE 446-1C: Lewy bodies (arrows) — eosinophilic intracytoplasmic inclusions with a dense core and pale halo, within melanized dopamine neurons of the SNc.
Three hallmark pathological features:
- Degeneration of dopaminergic neurons in the SNc (substantia nigra pars compacta)
- Reduced striatal dopamine
- Lewy pathology — intraneuronal alpha-synuclein aggregates as:
- Lewy bodies (in cell bodies)
- Lewy neurites (in axons)
Nondopaminergic involvement (explains nonmotor features):
Lewy pathology also affects:
- Cholinergic neurons of nucleus basalis of Meynert (NBM) → dementia, cognitive dysfunction
- Norepinephrine neurons of locus coeruleus (LC) → depression, sleep
- Serotonin neurons of raphe nuclei → mood disorders
- Olfactory system → anosmia
- Dorsal motor nucleus of vagus → autonomic dysfunction
- Peripheral autonomic nervous system, GI tract → constipation, gastroparesis
- Spinal cord and cerebral hemispheres
3. ETIOLOGY AND PATHOGENESIS
A. Genetics (TABLE 446-4 — Harrison's)
| Gene | Inheritance | Key Features |
|---|---|---|
| SNCA (alpha-synuclein) | AD | Median AAO 46 yrs; duplications cause classical PD; triplications cause severe, early-onset PD with dementia. Alpha-syn = major component of Lewy bodies |
| LRRK2 | AD | Most common known genetic form. Median AAO 56 yrs; clinically typical PD; slightly slower progression. p.G2019S mutation most common — founder effect in Ashkenazi Jewish and North African Berber populations |
| VPS35 | AD | Median AAO 52 yrs; clinically typical PD; very rare |
| GBA1 | Risk factor | Heterozygous variants = most common genetic risk factor; faster progression + greater cognitive risk |
| PRKN (Parkin) | AR | Most common AR cause; early-onset PD (median AAO 32 yrs, range 9-67 yrs) |
| PINK1 | AR | Mitochondrial kinase; early-onset |
| PARK7 (DJ-1) | AR | Very rare; early-onset |
"Double-hit" hypothesis (Harrison's 22e): Many PD cases may result from (1) genetic susceptibility factor + (2) toxic environmental exposure. Neither alone is sufficient.
Important genetic concept: Duplication/triplication of wild-type SNCA alone causes PD - meaning increased production of the normal protein is sufficient to cause disease.
Alpha-synuclein as a prion: Lewy pathology was found in healthy embryonic dopamine neurons transplanted into PD patients - implying the abnormal protein transferred from diseased to healthy cells. This suggests PD may be a prion-like disorder.
B. Environmental Factors
- MPTP (methyl-phenyl-tetrahydropyridine) — contaminant of illicit synthetic heroin; caused acute PD syndrome in California addicts in 1980s
- MPTP → oxidized (by MAO-B) to MPP+ → mitochondrial Complex I inhibitor → selective dopamine neuron death
- Importantly: MPTP/MPTP-like compounds have NOT been linked to sporadic PD
- Risk factors (inconsistent evidence): pesticides, rural living, farming, well water, solvents
- Possible protective factors: caffeine, cigarette smoking, NSAIDs, calcium channel blockers
C. Pathogenetic Mechanisms
- Oxidative stress — free radical production from dopamine metabolism
- Mitochondrial dysfunction — Complex I deficiency in SNc
- Proteasomal/lysosomal failure — impaired clearance of alpha-synuclein aggregates
- Neuroinflammation — activated microglia in SNc
- Apoptosis — final common pathway
D. Braak Staging (Alpha-synuclein spread)
Lewy pathology begins in the olfactory system, GI tract, and dorsal vagal nucleus and spreads in a predictable, sequential (caudal-to-rostral) manner to reach the SNc (mid-stage) and then the cerebral hemispheres (late stage). This explains why:
- Anosmia, constipation, RBD precede motor symptoms by years to decades
- These are now classified as premotor PD, not just risk factors
- Concept of "body-first" vs "brain-first" PD based on where pathology initiates
4. CLINICAL FEATURES (TABLE 446-1 — Harrison's)
A. Cardinal Motor Features (TRAP)
| Feature | Details |
|---|---|
| Tremor (rest) | "Pill-rolling"; 4-6 Hz; suppressed by voluntary movement; asymmetric onset |
| Rigidity | Cogwheel or lead-pipe; throughout range of motion |
| Akinesia/Bradykinesia | Slowing of movement; the most disabling feature |
| Postural instability | Impaired postural reflexes; late feature; major cause of falls |
B. Other Motor Features
- Micrographia — progressively smaller handwriting
- Masked facies (hypomimia) — reduced facial expression
- Reduced eye blinking
- Drooling (sialorrhoea)
- Hypophonia — soft, monotone voice
- Dysphagia — oral, pharyngeal, esophageal
- Freezing of gait — sudden arrest of movement ("feet glued to the ground"); major cause of falls; may respond to sensory cues (marching, stepping over imaginary line, singing)
- Festinating gait — small, shuffling, rapid steps; anteropulsion; reduced arm swing; en bloc turning
- Retropulsion — backward stepping on pull test
C. Non-Motor Features
| System | Features |
|---|---|
| Autonomic | Orthostatic hypotension, constipation, urinary urgency, sexual dysfunction, seborrhea, sialorrhoea |
| Sleep | REM sleep behaviour disorder (RBD) — often premotor; fragmented sleep; excess daytime sleepiness |
| Sensory | Anosmia/hyposmia (early premotor), pain, restless legs |
| Mood | Depression (~50% of PD patients), anxiety, apathy, panic attacks |
| Cognitive | Mild executive dysfunction early; dementia in up to 80% ultimately — primarily affects executive function and attention; relative sparing of language, memory, calculation |
| Psychosis | Visual hallucinations (typically formed, nonthreatening); often medication-induced |
5. DIAGNOSIS
Clinical Criteria
Historically: 2 of 3 features (tremor, rigidity, bradykinesia) → 24% error rate at autopsy
UK Brain Bank Criteria (>90% pathological accuracy):
- Core: Bradykinesia + rigidity
- Plus: rest tremor, asymmetry of onset, good response to levodopa
MDS Clinical Diagnostic Criteria (most current - Harrison's 22e):
- Core: Motor parkinsonism (bradykinesia + rigidity/tremor)
- Three additional categories:
- Supportive criteria (increase diagnostic confidence)
- Absolute exclusion criteria
- Red flags (must be counterbalanced by supportive criteria)
- Two certainty levels: Clinically Established PD and Clinically Probable PD
Supportive Criteria for PD (Harrison's)
- Clear beneficial response to dopaminergic therapy
- Rest tremor
- Anosmia
- Cardiac sympathetic denervation on MIBG scintigraphy
Red Flags (suggest atypical parkinsonism):
- Early postural instability or falls (within first 3 years)
- Early dementia (within first 1 year = DLB)
- Absence of rest tremor
- Symmetrical onset
- Poor or no response to adequate levodopa trial
- Early prominent autonomic failure
- Supranuclear gaze palsy (downward gaze palsy → PSP)
- Cerebellar signs
- Pyramidal signs
Investigations
DaT PET/SPECT:
- Shows reduced and asymmetric striatal dopamine transporter uptake
- Most affected: posterior putamen (with relative sparing of caudate)
- Does NOT reliably distinguish PD from atypical parkinsonism (both show reduced DaT)
FDG-PET (basal ganglia metabolic imaging):
- PD: decreased GPI activity + increased thalamic activity
- Atypical parkinsonism: reverse pattern
MRI: Typically normal in idiopathic PD; used to exclude structural causes
Biomarkers (Harrison's 22e - cutting edge):
- Alpha-synuclein seed amplification assay (SAA) — detects misfolded alpha-syn in CSF/skin/olfactory epithelium; highly sensitive and specific; increasingly used for early/premotor diagnosis
6. DIFFERENTIAL DIAGNOSIS
Classification of Parkinsonism (TABLE 446-2 — Harrison's)
| Category | Examples |
|---|---|
| PD | Sporadic; Genetic; PD with dementia/DLB |
| Atypical parkinsonism | MSA (MSA-p, MSA-c); PSP (parkinsonian form, Richardson form); Corticobasal syndrome (CBS) |
| Secondary parkinsonism | Drug-induced; Vascular; Tumor; Infection; NPH; Toxins (MPTP, CO, manganese) |
| Other neurodegenerative | Wilson's disease; Huntington's (Westphal variant); Spinocerebellar ataxias; PANK-associated neurodegeneration |
Key Distinguishing Features
| Diagnosis | Key Clue |
|---|---|
| Drug-induced | Neuroleptics, metoclopramide, flunarizine, cinnarizine, amiodarone, lithium, tetrabenazine |
| MSA | Parkinsonism + cerebellar signs + severe autonomic failure; alpha-syn in oligodendrocytes (GCIs); MRI: putaminal rim / hot cross bun sign |
| PSP | Early falls + downward gaze palsy + eyelid apraxia; tau pathology; MRI: hummingbird sign (midbrain atrophy) |
| CBS | Alien limb, apraxia, cortical sensory loss; asymmetric |
| DLB | Dementia develops before or within 12 months of parkinsonism; core: fluctuating cognition + recurrent visual hallucinations + RBD; GBA1 a major risk |
| Wilson's disease | Age <40; liver disease; Kayser-Fleischer rings; MUST exclude — treatable |
| DRD | Age <20; levodopa-responsive; no fluorodopa PET abnormality; no drug-induced dyskinesias; GTP-CH1 mutation |
7. TREATMENT
A. LEVODOPA — The Gold Standard
"Levodopa remains the most effective symptomatic treatment for PD and the gold standard against which new therapies are compared." — Harrison's 22e
History: Carlsson (1950s) → reserpine blocks dopamine → parkinsonism → reversed by levodopa. Hornykiewicz → striatal dopamine deficiency in PD → basis of replacement therapy.
Mechanism: Levodopa (DA precursor) crosses BBB → converted to dopamine in surviving SNc neurons → restores striatal DA.
Always combined with peripheral decarboxylase inhibitor:
- Carbidopa (USA) — brand: Sinemet
- Benserazide (Europe) — brand: Madopar
- Purpose: block peripheral levodopa conversion → prevent nausea, vomiting, orthostatic hypotension (from area postrema dopamine receptors that lie outside the BBB)
Available formulations (Harrison's 22e):
| Formulation | Example |
|---|---|
| Standard oral | Sinemet (carbidopa/levodopa) |
| Controlled-release | Sinemet CR / Madopar HP |
| Combined with COMT inhibitor | Stalevo (levodopa + carbidopa + entacapone) |
| Long-acting oral | Rytary |
| Intestinal gel infusion (continuous) | Levodopa-carbidopa intestinal gel |
| Subcutaneous continuous infusion | Available |
| Inhaled (pulmonary alveoli) | Inbrija — on-demand for off periods |
Motor Complications of Chronic Levodopa
Occur in most patients with ongoing disease progression:
-
Wearing-off effect
- Duration of benefit per dose shortens progressively
- Initial "long-duration response" (hours/days) → eventually approaches drug half-life (60-90 min)
- Due to loss of dopaminergic storage capacity as neurons degenerate
-
On-off phenomenon
- Rapid, unpredictable switches: "on" (drug working) ↔ "off" (parkinsonism returns)
- Patients may remain in "off" state for hours despite a levodopa dose
-
Dyskinesias
- Involuntary choreiform/dystonic movements
- Typically at peak dose (peak-dose dyskinesias)
On-demand therapies for off periods (3 FDA-approved):
- Inhaled levodopa (Inbrija) — fastest acting
- Subcutaneous apomorphine injection (Apokyn)
- Sublingual apomorphine (Kynmobi)
B. MAO-B INHIBITORS
| Drug | Notes |
|---|---|
| Selegiline | First-line option; DATATOP study: delayed need for levodopa |
| Rasagiline | ADAGIO study: 1 mg/d showed disease-modifying consistent benefit (delayed-start design); 2 mg dose failed (reason unclear) |
Mechanism of neuroprotection rationale: MAO-B inhibitors block oxidative conversion of MPTP → MPP+; also reduce dopamine oxidative metabolism; propargyl ring provides antiapoptotic effects in lab models.
C. DOPAMINE AGONISTS
| Drug | Type | Notes |
|---|---|---|
| Pramipexole | Non-ergot | Preferred; also helps depression |
| Ropinirole | Non-ergot | Preferred |
| Rotigotine | Non-ergot (patch) | Transdermal delivery |
| Apomorphine | Non-ergot (SC/SL) | On-demand rescue for off periods |
| Bromocriptine | Ergot | Less used now |
Advantages over levodopa:
- Longer duration of action
- Lower risk of dyskinesias
- Used as initial monotherapy in younger patients to delay dyskinesias
Important adverse effects: Nausea, orthostatic hypotension, somnolence (sudden sleep attacks — important for driving), hallucinations, impulse control disorders (gambling, hypersexuality, binge eating — particularly with pramipexole/ropinirole)
D. COMT INHIBITORS
| Drug | Notes |
|---|---|
| Entacapone | Preferred; no hepatotoxicity; multiple daily dosing |
| Opicapone | Once daily; no hepatotoxicity; taken on empty stomach |
| Tolcapone | Risk of fulminant hepatic necrosis → reserved for refractory cases only; requires liver function monitoring |
Mechanism: Block COMT → reduce levodopa conversion to 3-O-methyldopa (which competes with levodopa for CNS transport) → increase brain dopamine → reduce wearing-off
E. AMANTADINE
- NMDA glutamate receptor antagonist (also mild dopaminergic + anticholinergic)
- Uses: mild early PD symptoms; crucially — treatment of levodopa-induced dyskinesias in advanced PD
F. ANTICHOLINERGICS
- Benztropine, trihexyphenidyl
- Primarily for tremor in younger patients
- Avoid in elderly — anticholinergic burden (confusion, memory impairment, urinary retention, constipation)
G. NEUROPROTECTION (Major unmet need — Harrison's 22e)
"A neuroprotective or disease-modifying therapy that slows or stops disease progression remains the major unmet therapeutic need."
Current investigational targets:
- Anti-alpha-synuclein therapies (prevent aggregation/accumulation)
- LRRK2 inhibitors
- GBA1/GCase enhancers
- GLP-1 agonists (anti-inflammatory, pro-mitochondrial) — results in double-blind trials have been inconsistent
H. SURGICAL TREATMENT
Historical evolution (Harrison's):
- Motor cortex lesions → improved tremor but caused motor deficits → abandoned
- VIM thalamotomy → reduced tremor, no hemiparesis, but no benefit for bradykinesia/rigidity
- Posteroventral pallidotomy (GPi lesion) → improved rigidity, bradykinesia, tremor + markedly reduced dyskinesias; but bilateral lesions → dysphagia, dysarthria, cognitive impairment
- STN lesions → anti-parkinsonian + reduced levodopa need, but risk of hemiballismus
Deep Brain Stimulation (DBS) — Current standard:
- Electrode implanted into target → connected to subcutaneous stimulator over chest wall
- Disrupts pathological neurophysiological signals without making a brain lesion
- Fully reversible and adjustable (voltage, frequency, pulse duration, electrode configuration)
- Suitable for bilateral procedures safely
DBS targets:
| Target | Best for |
|---|---|
| STN (subthalamic nucleus) | All cardinal features; allows levodopa dose reduction |
| GPi (globus pallidus interna) | All cardinal features + especially dyskinesias; less risk of depression vs STN |
| VIM thalamus | Tremor only |
Indications for DBS:
- Severe tremor refractory to medications
- Motor fluctuations / dyskinesias not controlled by drug adjustments
- Good cognitive function
- Good levodopa response (except for tremor)
DBS side effects:
- Surgical: hemorrhage, infarction, infection
- Hardware: lead displacement, lead break, skin ulceration
- Stimulation: ocular/speech abnormalities, paresthesias, depression, rarely suicidality
DBS does NOT prevent nondopaminergic progression (freezing, falls, dementia continue to evolve).
Focused Ultrasound (FUS) Thalamotomy: Non-invasive; approved for tremor-predominant PD.
8. MANAGEMENT OF NONMOTOR FEATURES (Harrison's 22e)
Harrison's explicitly states: "Nondopaminergic features (especially falls and dementia) are the primary source of disability and main reason for hospitalization and nursing home placement in the levodopa era."
| Problem | Management |
|---|---|
| Depression (~50%) | Pramipexole (also helps motor features); SSRIs; tricyclics; don't withhold in major depression |
| Anxiety | Better dopaminergic control; short-acting benzodiazepines |
| Psychosis | Step-down: stop anticholinergics → stop amantadine → stop DA agonists → reduce levodopa → add atypical antipsychotic |
| Atypical antipsychotics | Clozapine (effective but needs CBC monitoring for agranulocytosis); Quetiapine (used but not proven in RCTs); Pimavanserin (5-HT2A inverse agonist; no motor worsening; mild efficacy; QT prolongation risk) |
| Dementia (PDD) | Rivastigmine (ChEI — approved for PDD); ultimately affects up to 80% |
| REM Sleep Behaviour Disorder | Clonazepam 0.5-1 mg at bedtime; melatonin; polysomnography if needed |
| Constipation | Osmotic laxatives, fibre, hydration; correlates with disease severity |
| Orthostatic hypotension | Fludrocortisone, midodrine, droxidopa, compression stockings |
| Sialorrhoea | Botulinum toxin injections into salivary glands |
| Freezing | Sensory cues (marching in place, stepping over imaginary line, counting); "on" period freezing has no specific treatment |
| Excess daytime sleepiness | Sodium oxybate (Xyrem) being evaluated; patient counselling re: driving |
Light therapy (Harrison's 22e — new): Exposure to specific light frequencies restores normal circadian rhythm (altered in PD) → both motor and nonmotor benefits, especially sleep and mood.
9. CURRENT MANAGEMENT ALGORITHM (Harrison's 22e)
DIAGNOSIS OF PD
|
├── No functional disability → MAO-B inhibitor (rasagiline/selegiline)
| [potential neuroprotection + mild symptom relief]
|
├── Functional disability present:
| |
| ├── Age <60 / Cognitively intact → DA AGONIST first
| | (delays dyskinesias; longer duration of action)
| |
| └── Age >70 / Cognitively impaired → LEVODOPA first
| (most effective; agonists poorly tolerated in elderly)
|
├── Motor complications develop (wearing-off/dyskinesias):
| → Adjust levodopa dosing/intervals
| → Add COMT inhibitor (entacapone/opicapone) for wearing-off
| → Add amantadine for dyskinesias
| → On-demand therapies for off periods (inhaled levodopa, apomorphine)
|
└── Refractory motor complications → Consider DBS (STN or GPi)
Key principle from Harrison's: Striatal dopamine terminal degeneration may be complete within 4 years of diagnosis — limiting potential benefit of neuroprotective therapy started late.
10. PROGNOSIS
- Progression typically over 5-15+ years
- Nondopaminergic features (dementia, falls, autonomic failure) cause the most late disability
- Dementia ultimately affects up to 80% of patients
- Leading cause of death: aspiration pneumonia; complications of immobility; falls
11. KEY EXAM FACTS AT A GLANCE
| Point | Fact |
|---|---|
| Pathological hallmark | Lewy bodies (alpha-synuclein) in SNc |
| Most common genetic form | LRRK2 (AD) |
| Most common AR form | PRKN/Parkin |
| Most common genetic risk factor | GBA1 (heterozygous) |
| Drug that causes parkinsonism + watch for | Metoclopramide (a neuroleptic used for GI) |
| Must always rule out in young-onset | Wilson's disease (treatable!) |
| Gold standard treatment | Levodopa |
| Most effective for dyskinesias | Amantadine; GPi-DBS |
| Antipsychotic that doesn't worsen parkinsonism | Pimavanserin (5-HT2A inverse agonist) |
| RBD treatment | Clonazepam 0.5-1 mg at bedtime |
| DBS preferred for dyskinesias | GPi (less depression risk vs STN) |
| Primary disability cause in levodopa era | Falls + dementia (nondopaminergic) |
| Premotor markers | Anosmia, constipation, RBD |
| Alpha-synuclein prion evidence | Lewy bodies found in transplanted fetal neurons from PD patients |
Source: Harrison's Principles of Internal Medicine, 22nd Edition (2025), Chapter 446: Parkinson's Disease and Related Disorders — C. Warren Olanow, Anthony H.V. Schapira, Christine Klein.
This is a shared conversation. Sign in to Orris to start your own chat.