Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathogenesis mechanism alveolar damage
Reading File
Reading File
Reading File
I now have comprehensive source material. Let me synthesize everything into a detailed, well-structured answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition (Berlin 2012)
ARDS is defined by:
- Acute onset within 7 days of a known clinical insult
- Bilateral opacities on chest imaging not explained by effusions, collapse, or nodules
- Respiratory failure not fully explained by cardiac failure or fluid overload
- PaO₂/FiO₂ < 300 with PEEP ≥ 5 cm H₂O (mild: <300; moderate: <200; severe: <100)
Triggers: Direct vs. Indirect Lung Injury
| Direct (Pulmonary) | Indirect (Non-Pulmonary) |
|---|---|
| Pneumonia (bacterial, viral, COVID-19) | Sepsis |
| Aspiration | Major trauma |
| Pulmonary contusion | Multiple transfusions |
| Toxic inhalation | Pancreatitis |
| Near-drowning | Cardiopulmonary bypass |
| Reperfusion injury | Drug overdose |
The final common pathway is the same regardless of etiology: diffuse alveolar damage (DAD).
Core Pathophysiological Mechanism
1. Breakdown of the Alveolar–Capillary Barrier
Under normal conditions, the alveolar–capillary membrane maintains a tight seal preventing fluid leak. In ARDS, damage to both the microvascular endothelium and the alveolar epithelium causes increased permeability. Unlike cardiogenic pulmonary edema (driven by elevated hydrostatic pressure), the edema fluid in ARDS is protein-rich (exudative), reflecting loss of barrier integrity rather than pressure excess.
- Epithelial injury — type I pneumocytes (which cover ~95% of the alveolar surface) are highly vulnerable to necrosis and apoptosis. Their loss disrupts barrier integrity and impairs alveolar fluid clearance.
- Endothelial injury — loss of pulmonary microvascular endothelial integrity is both necessary and sufficient for ARDS development. Mechanisms include cell necrosis, apoptosis, coagulation-related damage, and mechanical stretch.
2. Neutrophil Recruitment and Activation
Neutrophils are central mediators. In ARDS, BAL fluid shows a dramatic increase in neutrophil concentration (>80% of cells vs. <5% normally). The sequence:
- Initiating insult (e.g., sepsis, pneumonia) triggers systemic or local release of pro-inflammatory cytokines — primarily TNF-α, IL-1β, IL-6, IL-8, and complement fragments (C5a).
- Cytokine gradient drives neutrophil margination and sequestration within pulmonary capillaries and alveolar spaces.
- Neutrophil activation leads to release of:
- Proteases (elastase, collagenase, gelatinase) — degrade the extracellular matrix and alveolar epithelial proteins
- Reactive oxygen species (ROS) — oxidative damage to cell membranes
- Platelet-activating factor (PAF) and leukotrienes — amplify vascular permeability
- Neutrophil extracellular traps (NETs) — contribute to microvascular occlusion
Notably, ARDS can develop in severe neutropenia (e.g., chemotherapy patients), and lung injury appears more severe upon neutrophil recovery — suggesting neutrophils amplify but do not solely initiate injury.
3. Surfactant Dysfunction
Normal surfactant (dipalmitoylphosphatidylcholine + phosphatidylglycerol) reduces alveolar surface tension and prevents collapse. In ARDS:
- Decreased production by injured type II pneumocytes
- Protein inactivation — plasma proteins leaking into alveoli directly inhibit surfactant function
- Neutrophil elastase degrades surfactant protein A
- Shift from large (active) to small (inactive) surfactant aggregates
The net result is diffuse alveolar collapse (microatelectasis) and reduced compliance. This distinguishes adult ARDS from neonatal RDS, where surfactant deficiency is the primary cause rather than a secondary consequence.
4. Coagulation Cascade Activation
Alveolar flooding triggers the coagulation system:
- Procoagulant mediators (tissue factor, PAI-1) are elevated in BAL fluid
- Fibrin deposition within alveolar spaces and pulmonary microvasculature contributes to hyaline membrane formation
- Microvascular thrombosis reduces perfused capillary surface area, worsening hypoxemia and increasing dead space
- Activated protein C (an endogenous anticoagulant) is reduced; its anti-inflammatory effects (downregulating IL-6, attenuating neutrophil activation) are impaired
5. Impaired Alveolar Fluid Clearance
Normally, Na⁺ channels on type II alveolar epithelial cells actively transport sodium from alveolar fluid → cytoplasm, creating an osmotic gradient that draws water out. In ARDS:
- Hypoxia downregulates apical Na⁺ channel (ENaC) expression and activity
- Hypoxia also impairs basolateral Na⁺/K⁺-ATPase activity
- Elevated nitric oxide disrupts β-adrenergic receptor signaling, blunting catecholamine-stimulated fluid clearance
The result: protein-rich edema accumulates and persists rather than being actively cleared.
Phases of ARDS (Histological Correlation)
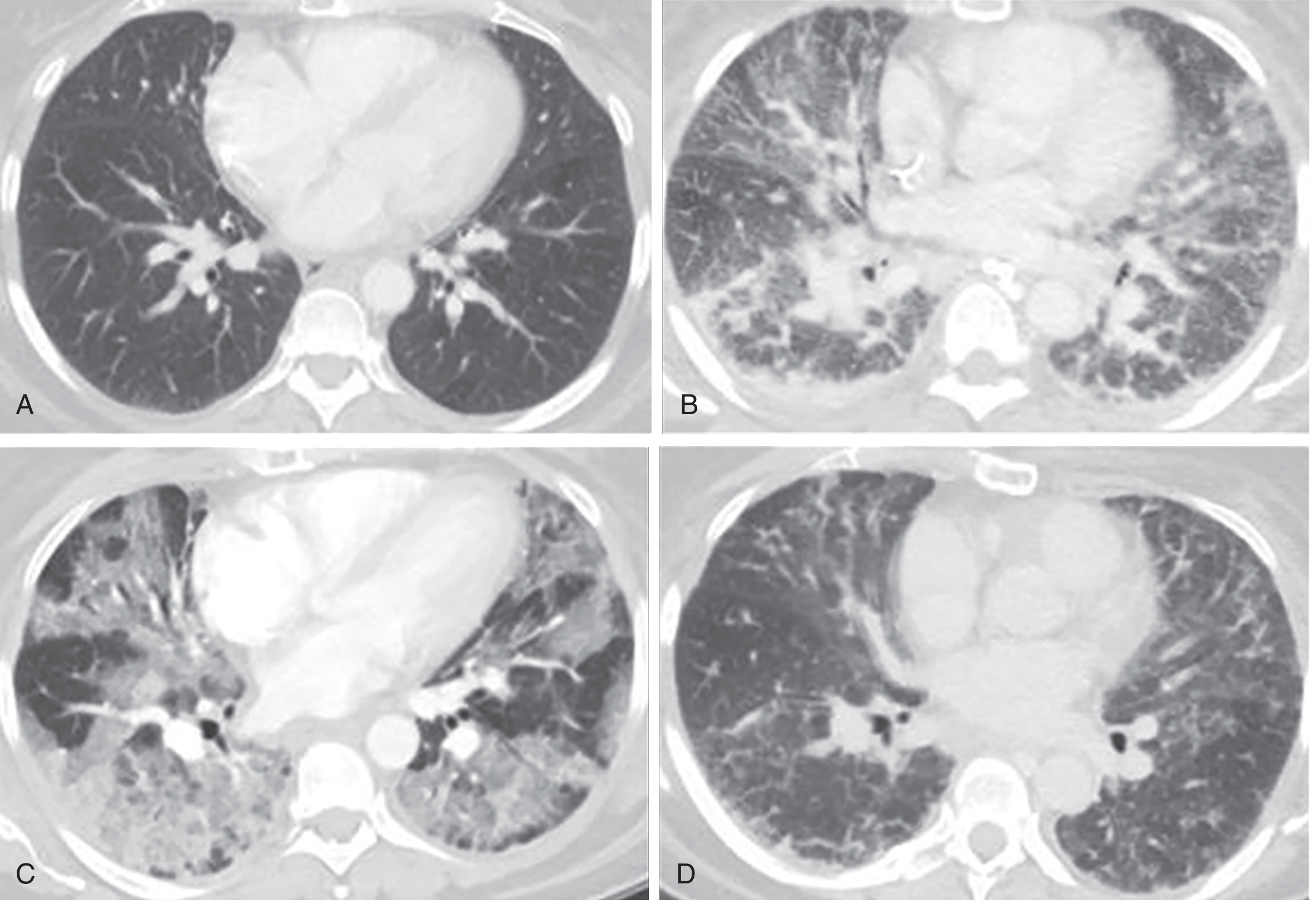
CT correlation of the three phases of ARDS (Murray & Nadel's)
| Phase | Timing | Pathology |
|---|---|---|
| Exudative | Days 1–7 | Hyaline membranes (protein + cellular debris + surfactant), protein-rich alveolar flooding, neutrophil infiltration, type I pneumocyte necrosis → DAD |
| Proliferative | Days 7–21 | Hyaline membrane reabsorption, type II pneumocyte hyperplasia, fibroblast proliferation, early collagen deposition, decreased neutrophils |
| Fibrotic | > Day 21 | Pulmonary fibrosis (subset of patients), obliteration of pulmonary capillaries, interstitial/alveolar collagen deposition |
Elevated N-terminal procollagen peptide III (a marker of collagen synthesis) can be detected in BAL as early as 24 hours after the onset of ARDS, indicating fibrotic remodeling begins almost immediately.
Diffuse Alveolar Damage: Histology and Imaging
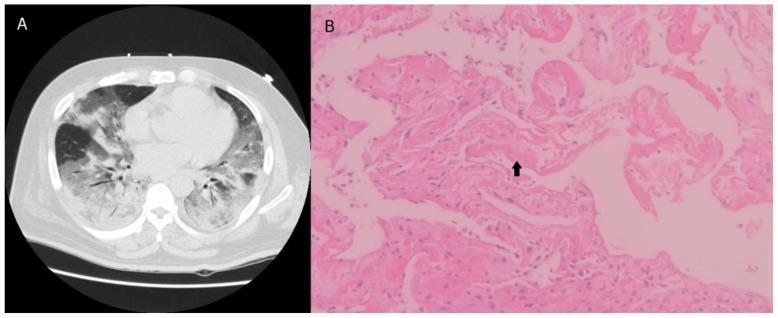
DAD: CT showing bilateral ground-glass opacities + histology showing hyaline membrane (arrow) and thickened alveolar walls
Pulmonary Physiology: How Damage Causes Hypoxemia
| Mechanism | Effect |
|---|---|
| Right-to-left intrapulmonary shunt | Collapsed/fluid-filled alveoli are perfused but not ventilated → deoxygenated blood enters circulation |
| Low V/Q areas | Partially flooded alveoli ventilated but poorly — worsens oxygenation |
| Increased dead space | Pulmonary microvascular occlusion perfuses poorly → elevated V/Q → CO₂ retention despite normal PaCO₂ initially |
| Decreased compliance | Heavy, edematous lung ("wet sponge") → reduced lung volumes, FRC, and increased work of breathing |
| Pulmonary hypertension | Hypoxic vasoconstriction + intravascular fibrin deposition + compression by positive-pressure ventilation |
Genetic and Host Factors
- ACE2 — SARS-CoV-1/2 infection via ACE2 reduces its surface expression, removing its protective effect on the lung (ACE2 normally degrades angiotensin II, which promotes inflammation and permeability)
- Surfactant protein gene variants and other SNPs modify susceptibility
- Angiopoietin-2 (released by injured endothelium) disrupts endothelial barrier; elevated levels predict ARDS development and severity
Summary Cascade
Inciting insult (sepsis, pneumonia, trauma, etc.)
↓
Systemic/local cytokine storm (TNF-α, IL-1β, IL-6, IL-8)
↓
Neutrophil sequestration + activation in pulmonary capillaries
↓
Protease + ROS release → endothelial + epithelial injury
↓
Alveolar-capillary barrier breakdown
↓
Protein-rich exudate floods alveoli
↓
Surfactant dysfunction → microatelectasis
Impaired Na⁺/fluid clearance → edema persists
Fibrin deposition → hyaline membranes + microvascular occlusion
↓
V/Q mismatch + shunt + decreased compliance
↓
Refractory hypoxemia (hallmark of ARDS)
↓
Proliferative → Fibrotic phase (in survivors/non-resolvers)
Sources: Murray & Nadel's Textbook of Respiratory Medicine (Chapter 134); Goldman-Cecil Medicine (Chapter 90/91); Sabiston Textbook of Surgery (Chapter on Critical Care)
This is a shared conversation. Sign in to Orris to start your own chat.