Albinism
Reading File
Finding Sources
Searching PubMed
albinism melanin genetics
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
I now have all the material needed for a comprehensive, well-illustrated answer.
Albinism
Albinism is a genetically heterogeneous group of disorders characterized by reduced or absent melanin production in the skin, hair, and/or eyes. It results from defects in melanin biosynthesis - primarily in the tyrosinase pathway.
Biochemical Basis
Melanin is synthesized from tyrosine in melanocytes within organelles called melanosomes. The key enzyme is tyrosinase (a copper-requiring enzyme), which catalyzes the first two steps in the conversion of tyrosine to melanin. Defects in this enzyme, or in proteins required for melanosome biogenesis, trafficking, and structure, result in albinism.
The pathway:
Tyrosine → (tyrosinase) → DOPA → (tyrosinase) → DOPAquinone → ... → Melanin
- Lippincott's Biochemistry, 8th ed., p. 769
Classification
Albinism is broadly divided into two types:
| Type | Structures affected |
|---|---|
| Oculocutaneous Albinism (OCA) | Eyes + skin + hair |
| Ocular Albinism (OA) | Eyes only |
Most forms are autosomal recessive; ocular albinism is commonly X-linked.
Oculocutaneous Albinism (OCA) - The 7 Types
OCA1 - Tyrosinase Gene Mutations
- Gene: TYR (chromosome 11q14) - encodes tyrosinase
- Prevalence: ~40% of OCA worldwide; most common in Japanese and Europeans (~1:40,000)
- Two subtypes:
- OCA1A: Complete absence of tyrosinase activity. White hair, white skin (does not tan), pink/translucent irises. VA decreased to 20/400. The most severe form.
- OCA1B ("yellow mutant"): Residual tyrosinase activity. Pigment can develop with age (from age 1-3 years); patients can tan. A temperature-sensitive variant (OCA1-TS) exists where the enzyme works only below 37°C - patients develop dark hair in cooler acral areas at puberty.
OCA2 - Most Common Worldwide
- Gene: OCA2 (formerly P-gene, chromosome 15q12) - encodes an integral melanosomal membrane protein important for tyrosine transport and melanosomal biogenesis
- Prevalence: ~50% of OCA worldwide; up to 1:4,000 in parts of Africa (most common form there)
- Formerly called "tyrosinase-positive" or "brown OCA"
- Newborns have pigmented hair; nevi and freckles are common; pink irises not usually seen
- Pigmentation and visual acuity improve from infancy through adolescence
- Important association: Deletions in the chromosomal region adjacent to the OCA2 gene cause Prader-Willi and Angelman syndromes
OCA3 - "Rufous" Albinism
- Gene: TYRP1 (chromosome 9p23) - tyrosinase-related protein 1, a structural cofactor for tyrosinase
- Primarily seen in Black African patients
- Patients have red hair and reddish-brown skin (hence "rufous")
- Visual abnormalities may not be detectable
OCA4
- Gene: SLC45A2 (MATP gene) - membrane-associated transporter protein
- Phenotypically identical to OCA2; variable hypopigmentation; nystagmus in many
OCA5
- Mapped to chromosome 4q24
- Described in a Pakistani family: golden hair, white skin, nystagmus, photophobia, impaired VA
OCA6
- Gene: SLC24A5 - solute carrier protein important in melanosomal architecture
- Found in diverse ethnicities; hair color ranges from white to dark brown
OCA7
-
Gene: C10orf11 - member of the leucine-rich repeat proteins
-
Andrews' Diseases of the Skin, pp. 1009-1010; Kanski's Clinical Ophthalmology 10th ed., p. 662; Emery's Medical Genetics, p. X
Ocular Features (Common to All Forms)
The ocular phenotype is the most consistent finding across all types of albinism, because melanin plays a critical developmental role in the eye.
Visual acuity: Severely reduced - typically < 6/60 (20/200 equivalent) in OCA1A, due to foveal hypoplasia (the fovea fails to develop properly without melanin signals during embryogenesis)
Nystagmus: Typically pendular and horizontal; worsens in bright light; lessens with age
Iris: Diaphanous and translucent ("pink-eyed" appearance) - in severe forms, the choroidal vasculature is visible through the iris, giving a red/pink color
Fundus: Severe hypopigmentation; large choroidal vessels are conspicuous; absent foveal pit and poorly formed perimacular arcades (see fundus image below)
Optic chiasm: Abnormal decussation - more fibers cross than normal (majority from each eye cross to the contralateral hemisphere), detectable on visual evoked potentials (crossed asymmetric VEPs)
Other: High refractive errors, strabismus, absent stereopsis, photophobia
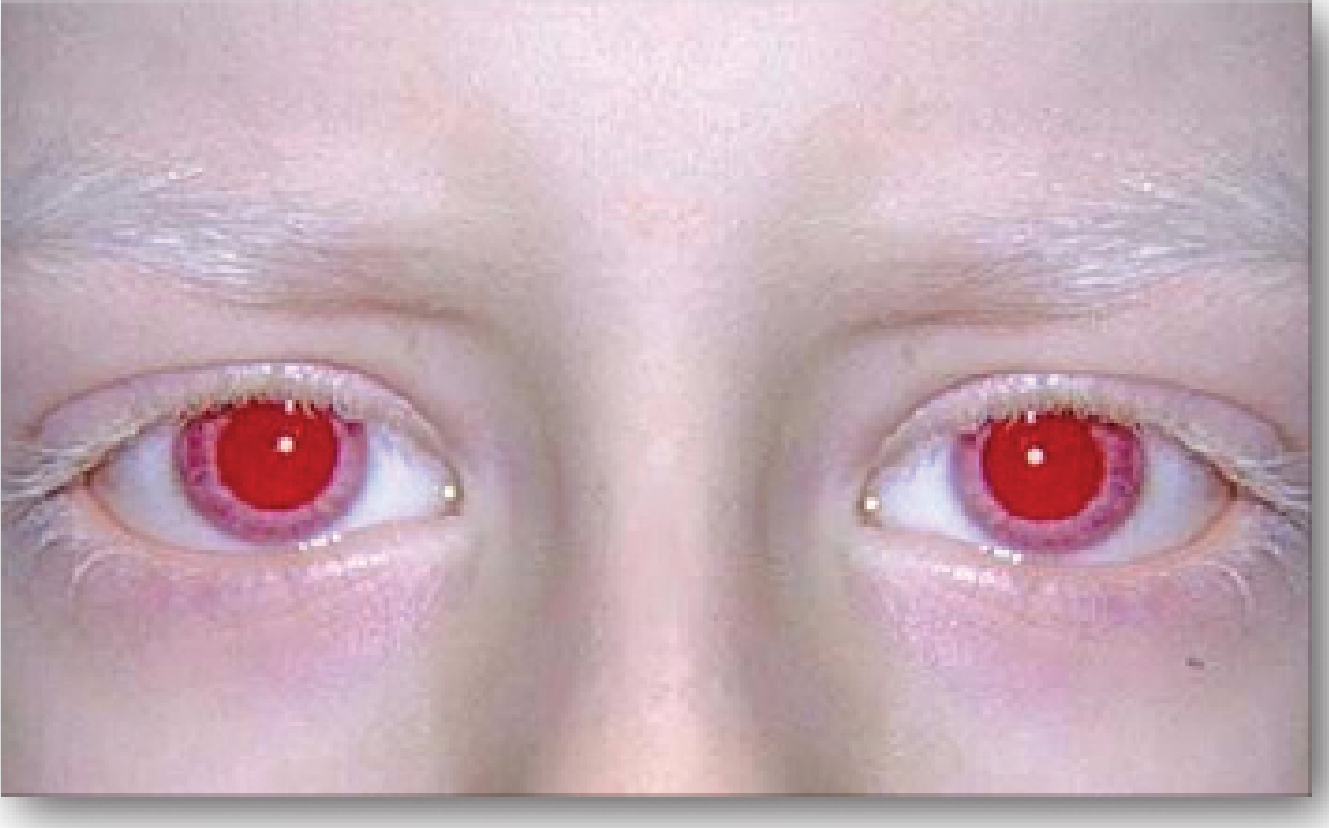
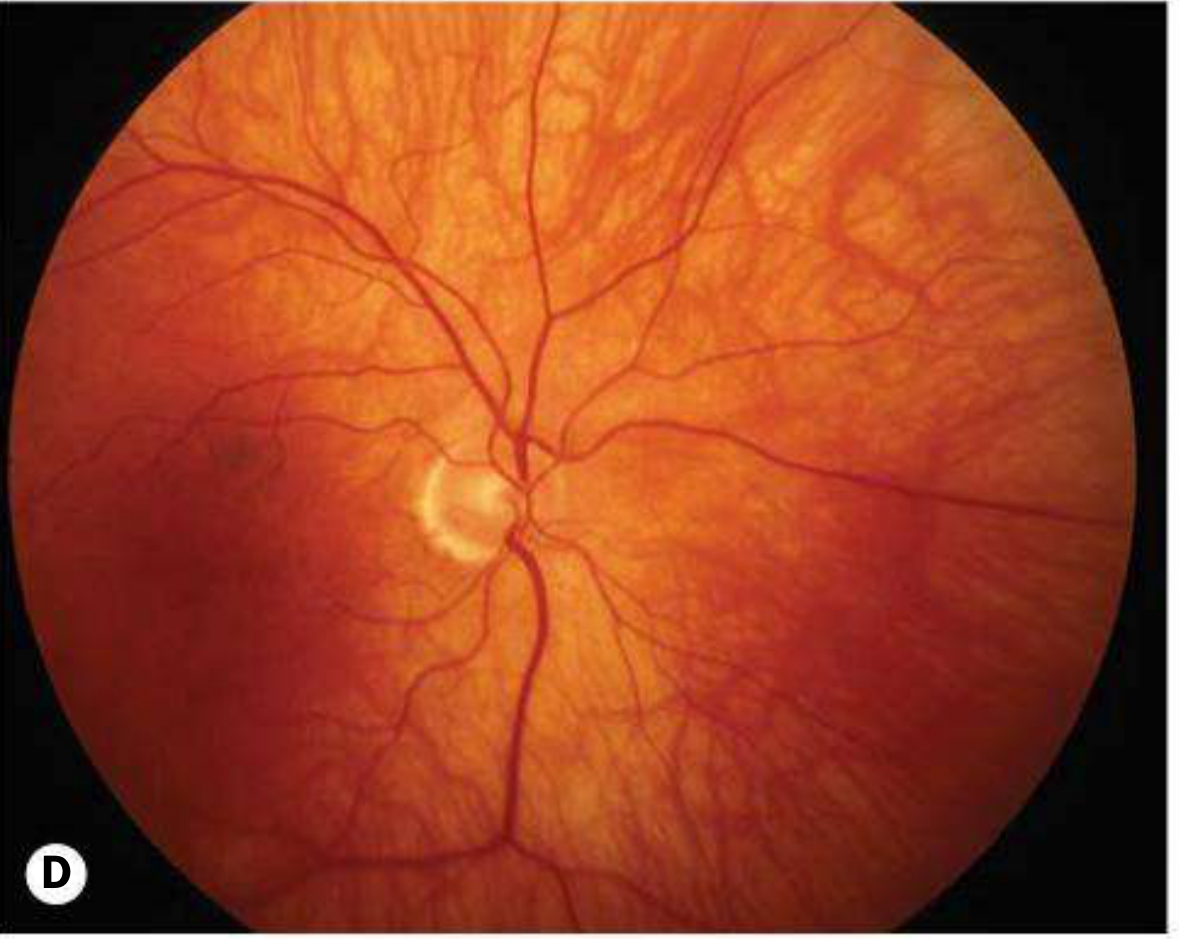
- Kanski's Clinical Ophthalmology 10th ed., pp. 662-664
Syndromic Associations
Several syndromes include albinism as a feature, with serious systemic consequences:
Hermansky-Pudlak Syndrome (HPS)
- Inheritance: Autosomal recessive; lysosomal storage disease
- Features: OCA + platelet dysfunction (impaired dense granules → easy bruising, excessive bleeding) + pulmonary fibrosis + granulomatous colitis
- Can be life-threatening
Chediak-Higashi Syndrome
- Inheritance: Autosomal recessive; mutation in LYST (lysosomal regulator protein)
- Features: OCA + failure of phagolysosome formation → recurrent pyogenic infections (especially Staphylococcus) + giant cytoplasmic granules in leukocytes + neurological deterioration
- Predisposes to accelerated lymphoma-like phase
Waardenburg Syndrome
-
Inheritance: Autosomal dominant
-
Features: White forelock, poliosis, synophrys (monobrow), sensorineural deafness, heterochromia iridis, sometimes limb abnormalities
-
Caused by mutations in PAX3, MITF, or SOX10 genes - not a true melanin synthesis defect but a melanocyte migration/development disorder
-
Kanski's Clinical Ophthalmology 10th ed., pp. 662-664; Andrews' Diseases of the Skin
Cutaneous Complications
- Skin cancer risk: Significantly increased risk of basal cell carcinoma and squamous cell carcinoma (due to lack of melanin photoprotection)
- Melanoma risk is also elevated
- Dermoscopy is challenging due to absent pigment; reflectance confocal microscopy may help
Management
| Concern | Intervention |
|---|---|
| Sun damage/skin cancer | Aggressive sun protection (SPF 50+ sunscreen, protective clothing, sun avoidance) |
| Vitamin D deficiency | Supplementation required due to sun avoidance |
| Visual impairment | Refractive correction; tinted lenses/sunglasses for photophobia |
| Nystagmus | May benefit from prism glasses; surgical options in select cases |
| Strabismus | Patching/surgery as appropriate |
| Skin surveillance | Regular dermatology review; biopsy any changing or atypical nevi |
There is no cure; treatment is supportive and preventive. Gene therapy research is ongoing but not yet in clinical use.
Summary Table: OCA Types
| Type | Gene | Chromosome | Key Features |
|---|---|---|---|
| OCA1A | TYR | 11q14 | Complete absence of melanin; most severe |
| OCA1B | TYR | 11q14 | Residual activity; pigment may develop |
| OCA2 | OCA2 (P-gene) | 15q12 | Most common worldwide; tyrosinase-positive |
| OCA3 | TYRP1 | 9p23 | Red/rufous phenotype; mainly African |
| OCA4 | SLC45A2 | 5p | Like OCA2; common in Japan |
| OCA5 | unknown | 4q24 | Pakistani families; golden hair |
| OCA6 | SLC24A5 | 15q21 | Variable; diverse ethnicities |
| OCA7 | C10orf11 | 10q22 | Leucine-rich repeat protein |
Sources:
- Andrews' Diseases of the Skin (Clinical Dermatology), pp. 1009-1010
- Kanski's Clinical Ophthalmology, 10th ed., pp. 662-664
- Emery's Elements of Medical Genetics and Genomics
- Lippincott Illustrated Reviews: Biochemistry, 8th ed., p. 769
- The Developing Human (Clinically Oriented Embryology)
This is a shared conversation. Sign in to Orris to start your own chat.