Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND mechanism
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have very detailed, comprehensive content from Murray & Nadel and Robbins on ARDS pathogenesis. I have enough to write a thorough, well-structured answer. Let me now compose the full response.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is a syndrome of noncardiogenic pulmonary edema driven by severe diffuse lung inflammation, increased alveolar-capillary permeability, and impaired gas exchange. No single pathophysiologic pathway explains all cases - it is best understood as a convergence of injurious processes triggered by diverse insults.
Definition and Diagnostic Criteria (Berlin 2012)
ARDS is defined by four criteria:
- Acute onset - within 1 week of a known clinical insult
- Bilateral opacities on chest imaging not fully explained by effusions, atelectasis, or nodules
- Non-cardiogenic - respiratory failure not explained by cardiac failure or fluid overload
- Hypoxemia stratified by PaO2/FiO2 ratio (with PEEP ≥5 cmH2O):
- Mild: 200-300 mmHg
- Moderate: 100-200 mmHg
- Severe: <100 mmHg
The most common triggers (>50% of cases) are sepsis, diffuse pulmonary infections, gastric aspiration, and mechanical trauma - (Robbins, Cotran & Kumar Pathologic Basis of Disease).
Histopathologic Correlate: Diffuse Alveolar Damage (DAD)
The pathologic hallmark of ARDS is diffuse alveolar damage (DAD), though only ~50% of ARDS patients confirmed on autopsy show DAD; the rest may have infectious pneumonia, diffuse alveolar hemorrhage, or eosinophilic pneumonia.
DAD progresses through three overlapping phases:
| Phase | Timing | Key Features |
|---|---|---|
| Exudative | Days 1-7 | Hyaline membrane formation, protein-rich alveolar edema, neutrophilic infiltration, type I pneumocyte necrosis |
| Proliferative | Days 7-21 | Hyaline membrane reorganization, type II pneumocyte proliferation, early fibrosis, reduced neutrophils |
| Fibrotic | >2-3 weeks | Pulmonary fibrosis, obliteration of capillaries, collagen deposition |
Notably, fibroproliferation may begin simultaneously with early inflammatory injury rather than strictly after it, as evidenced by elevated N-terminal procollagen peptide III in BAL fluid within the first 24 hours - (Murray & Nadel's Textbook of Respiratory Medicine).
Core Pathogenesis: Step-by-Step
The central event is injury to the alveolar-capillary membrane, which has two components: the alveolar epithelium and the microvascular endothelium. Damage to either (or both) breaks down the barrier, allowing fluid and inflammatory cells to flood the airspaces.
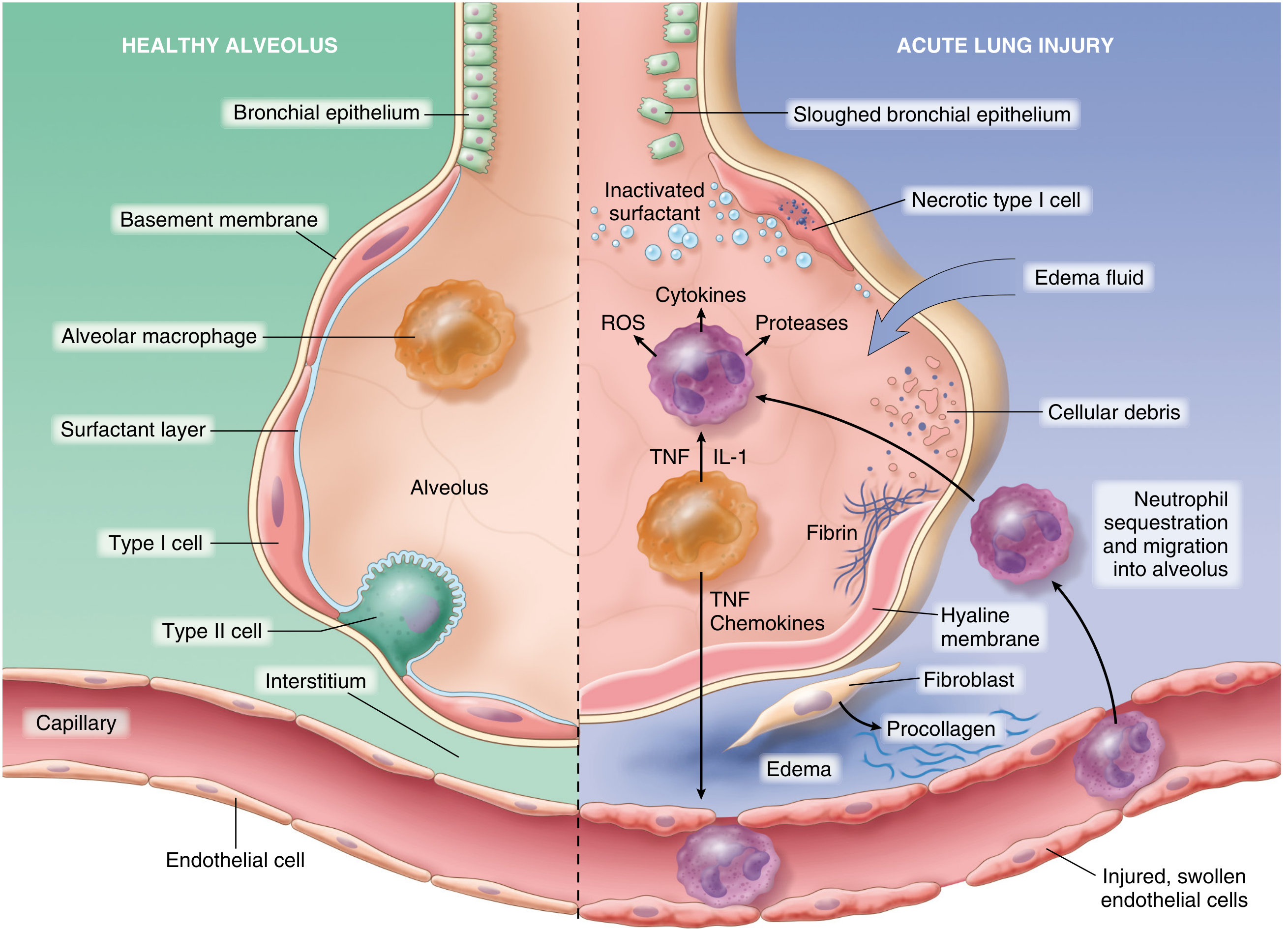
1. Initiating Injury - Two Entry Points
Direct (pulmonary) insults - pneumonia, aspiration, inhalation injury, pulmonary contusion - injure the alveolar epithelium first. Indirect (extrapulmonary) insults - sepsis, pancreatitis, major trauma, massive transfusion - reach the lung through the bloodstream and activate pulmonary endothelium directly.
In either pathway, resident alveolar macrophages sense the damage and release TNF-α, IL-1, and IL-8, which activate the neighboring endothelium and amplify the inflammatory cascade - (Robbins).
2. Endothelial Activation and Barrier Breakdown
Circulating inflammatory mediators (TNF, IL-1, endotoxin, complement fragments) activate pulmonary capillary endothelial cells, causing them to:
- Upregulate adhesion molecules (ICAM-1, VCAM-1, E-selectin, P-selectin)
- Express procoagulant proteins
- Release chemokines (IL-8, MCP-1) that attract and activate neutrophils
Loss of pulmonary vascular endothelial barrier integrity is both necessary and sufficient for ARDS to develop - (Murray & Nadel).
3. Neutrophil Sequestration and Activation - The Central Amplifier
Neutrophils play the dominant effector role:
- Mechanical trapping: Pulmonary capillaries are narrower than the average neutrophil diameter. Activated neutrophils become "stiff" due to actin cytoskeleton reorganization and cannot deform to pass through, leading to microvascular sequestration. This transient leukopenia is one of the earliest signs of ARDS.
- Migration into alveoli: Sequestered neutrophils can migrate into the interstitium even without the usual L-selectin/β2-integrin adhesion mechanism. They further destabilize endothelial barriers, promoting more neutrophil influx in a self-amplifying cycle.
- Degranulation and toxic release: Once in the alveoli, neutrophils release:
- Neutrophil elastase (NE) - degrades cadherins (adherens junction proteins), growth factors, and cytokines; its destruction of epithelial and endothelial cell-cell junctions promotes alveolar flooding
- Matrix metalloproteinases - further degrade the extracellular matrix
- Reactive oxygen species (ROS) - oxidative damage to lipids, proteins, and DNA
- Cytokines and chemokines - self-amplifying inflammatory loop
- Neutrophil Extracellular Traps (NETs) - directly contribute to lung damage - (Murray & Nadel; Robbins)
Neutrophil-platelet interactions also amplify activation via mutual signaling and NET formation.
Intracellular signaling pathways involved include p38 MAPK (activated by LPS, drives TNF-α and MIP-2 production) and PI3K-γ (drives neutrophil chemotaxis toward IL-8 and fMLP).
4. Alveolar Epithelial Injury
Type I pneumocytes (covering 95% of alveolar surface area) are particularly vulnerable and undergo necrosis and apoptosis. Their loss:
- Disrupts the physical barrier, allowing edema fluid into the airspace
- Impairs alveolar fluid clearance (normally driven by Na+/K+-ATPase on type II cells)
Type II pneumocytes are also damaged, leading to:
- Reduced surfactant production and secretion
- Loss of the regenerative capacity for the alveolar epithelium
5. Surfactant Dysfunction
Normal surfactant (produced by type II pneumocytes) reduces alveolar surface tension and prevents collapse. In ARDS:
- Protein-rich edema fluid inactivates surfactant by competing for the air-liquid interface
- Phospholipase A2 (elevated in pancreatitis-related ARDS and other triggers) enzymatically degrades surfactant phospholipids
- Total surfactant pool is reduced
- The result is alveolar collapse (atelectasis), reduced compliance, and worsened V/Q mismatch - (Murray & Nadel).
6. Coagulation and Fibrin Deposition
ARDS activates the coagulation cascade within the lung:
- Increased tissue factor expression on endothelium and macrophages
- Suppressed fibrinolysis (elevated PAI-1)
- Fibrin and cellular debris accumulate in alveolar spaces, forming the characteristic hyaline membranes
- Microvascular thrombi occlude capillaries, contributing to pulmonary hypertension and dead-space ventilation
7. Na+ and Water Transport Failure
Under normal conditions, alveolar fluid is cleared by active Na+ transport across the epithelium (via ENaC channels on type I/II cells, pumped out by Na+/K+-ATPase on type II cells). In ARDS:
- Epithelial damage reduces functional Na+ transport capacity
- Edema cannot be cleared even as hydrostatic pressures normalize
- Impaired fluid clearance correlates with worse outcomes
8. Angiopoietins - Vascular Instability
Angiopoietin-1 (Ang-1) stabilizes the vasculature by activating Tie-2 receptors on endothelium. Angiopoietin-2 (Ang-2) is released from Weibel-Palade bodies of activated endothelial cells and competitively antagonizes Ang-1/Tie-2 signaling, destabilizing the endothelium and promoting vascular leak. Elevated plasma Ang-2 levels are associated with worse outcomes in ARDS - (Murray & Nadel).
9. Ventilator-Induced Lung Injury (VILI)
Once mechanical ventilation is initiated, the heterogeneous distribution of DAD lesions (gravity-dependent consolidation vs. nearly normal nondependent zones) means that delivered tidal volumes are distributed unevenly:
- High-volume or high-pressure ventilation overdistends aerated regions (volutrauma/barotrauma)
- Cyclic opening and closing of collapsed alveoli causes atelectrauma
- These mechanical stresses trigger further cytokine release (biotrauma), which can spill into the systemic circulation and contribute to multi-organ dysfunction - (Goldman-Cecil Medicine).
V/Q Mismatch and Hypoxemia
The lesions are not evenly distributed: dependent zones are flooded/consolidated (perfused but not ventilated - true shunt), while nondependent zones may be nearly normal. This severe ventilation-perfusion (V/Q) mismatch is the primary mechanism of hypoxemia. Unlike simple V/Q mismatch, the hypoxemia in ARDS responds poorly to supplemental oxygen alone because true intrapulmonary shunt (zero ventilation) is not corrected by raising FiO2 - (Robbins; Murray & Nadel).
Resolution (or Failure to Resolve)
If the triggering insult resolves:
- Alveolar macrophages clear debris and release TGF-β and PDGF, stimulating fibroblast proliferation and collagen deposition
- Type II pneumocytes proliferate to reconstitute the alveolar lining
- Endothelial restoration occurs through uninjured capillary proliferation
In patients with persistent ARDS (>2 weeks), a fibroproliferative phase dominates, leading to pulmonary fibrosis and chronic lung function impairment. This is also when excess collagen synthesis can be detected in BAL fluid as early as day 1 of illness - suggesting fibrosis may run in parallel with inflammation, not just sequentially after it.
Summary Diagram of Pathogenesis
Insult (sepsis / aspiration / trauma / infection)
↓
Macrophage activation → TNF-α, IL-1, IL-8
↓
Endothelial activation → ↑ adhesion molecules, procoagulants, chemokines
↓
Neutrophil sequestration (microvascular stiffening)
↓
Neutrophil migration into alveoli
↓
Release of: ROS + NE + MMPs + cytokines + NETs
↓
┌────────────────────────────────────────┐
│ Epithelial necrosis (type I >> type II) │
│ ↓ surfactant production & function │
│ ↓ Na+/fluid clearance │
│ Hyaline membrane formation │
│ Fibrin deposition, microthrombosis │
└────────────────────────────────────────┘
↓
Alveolar flooding + atelectasis + ↑ dead space
↓
V/Q mismatch → refractory hypoxemia
↓
Mechanical ventilation → potential VILI (barotrauma/biotrauma)
Key References:
- Murray & Nadel's Textbook of Respiratory Medicine - Chapter 134: Etiology and Pathogenesis of ARDS
- Robbins, Cotran & Kumar - Pathologic Basis of Disease - Chapter 15: Acute Lung Injury and ARDS (Diffuse Alveolar Damage)
- Goldman-Cecil Medicine - Chapter 91: Mechanical Ventilation in ARDS
- Harrison's Principles of Internal Medicine 22E - Chapter on Sepsis-associated ARDS
This is a shared conversation. Sign in to Orris to start your own chat.