1. Describe bilirubin metabolism 2. Enumerate the etiology and pathogenesis of jaundice 3. Distinguish between direct and indirect hyperbilirubinemia.
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content from multiple authoritative textbooks. Let me compile the full, detailed answer.
Bilirubin Metabolism, Jaundice, and Hyperbilirubinemia
1. Bilirubin Metabolism
Source of Bilirubin
Bilirubin is a tetrapyrrole compound and the end product of heme catabolism. A healthy adult produces approximately 4 mg/kg/day (~250-300 mg/day in a 70-kg person).
- 70-80% derives from degradation of hemoglobin released from senescent erythrocytes (destroyed in the spleen, bone marrow, and liver Kupffer cells)
- 20-30% comes from breakdown of other heme-containing proteins (e.g., cytochrome oxidases, catalase) in hepatocytes, as well as ineffective erythropoiesis
Step 1: Heme to Bilirubin (in Peripheral Tissues)
The conversion of heme to bilirubin occurs in two sequential enzymatic steps:
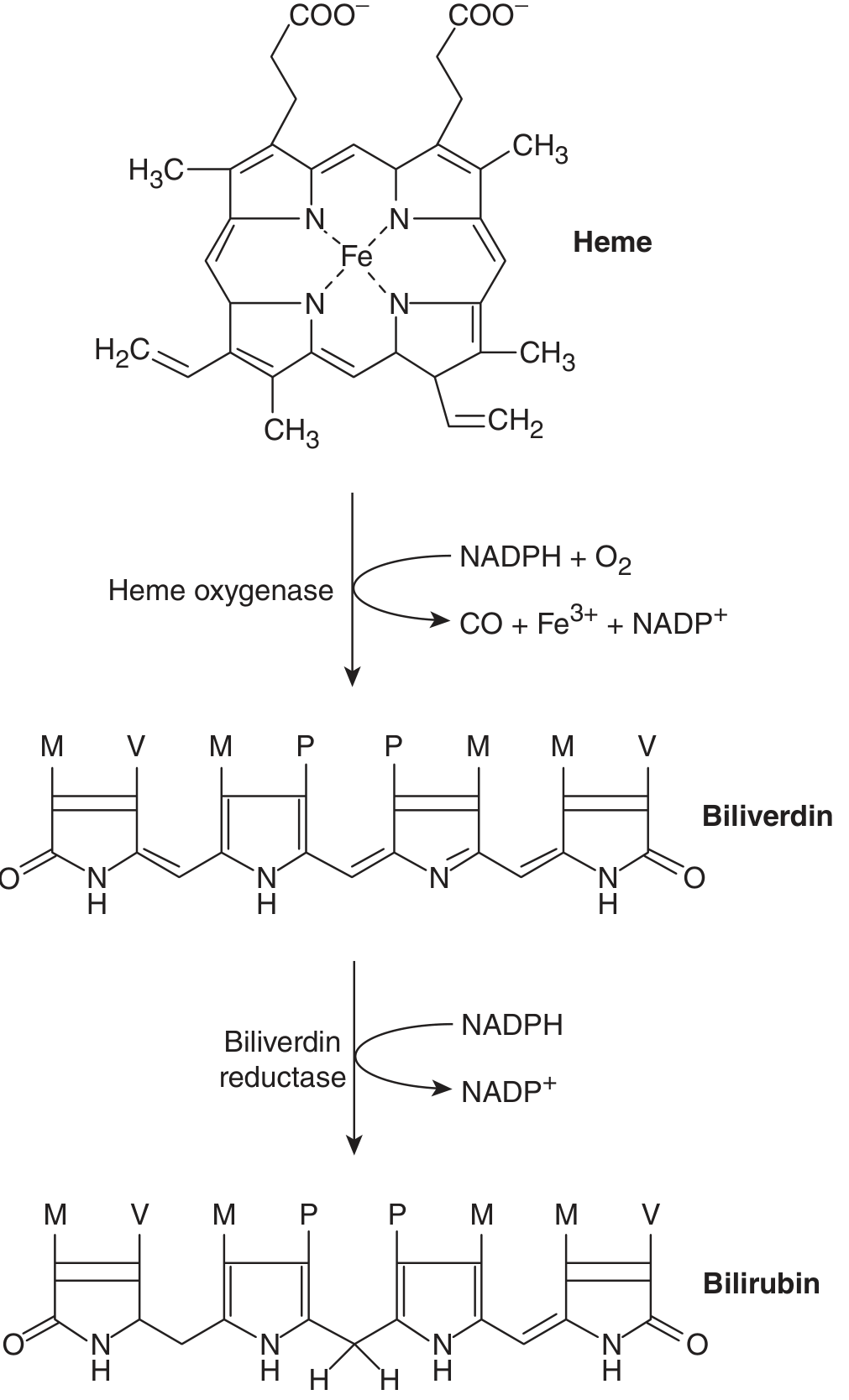
- Heme oxygenase (HO) opens the porphyrin ring of heme, releasing iron (Fe³+), carbon monoxide (CO), and producing biliverdin (a water-soluble green pigment). Two isoforms exist: HO-1 (inducible, ubiquitous) and HO-2 (constitutive, found in hepatocytes). The reaction requires NADPH and O₂.
- Biliverdin reductase (BVRA) then reduces biliverdin IXα to bilirubin IXα (the predominant form), using NADPH.
The resulting bilirubin is highly hydrophobic and water-insoluble in its unconjugated (free) form.
Step 2: Transport in Blood
Unconjugated bilirubin circulates in plasma tightly and non-covalently bound to albumin (due to its insolubility in aqueous solution). It cannot be filtered at the glomerulus and therefore does not appear in urine.
Step 3: Hepatic Handling - Four Key Steps
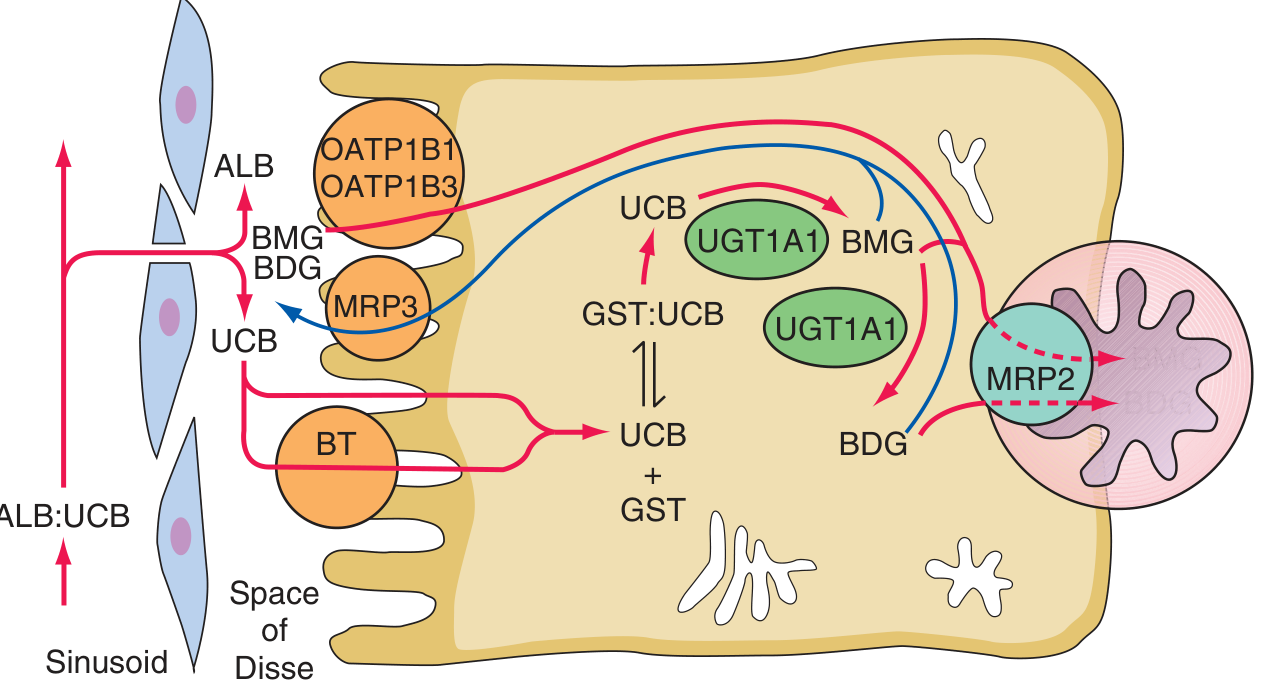
(a) Hepatocellular Uptake
Albumin-bound bilirubin passes through sinusoidal endothelial fenestrae to reach the hepatocyte surface. Free bilirubin is taken up via a proposed bilirubin transporter (BT) and possibly via organic anion transporting polypeptides (OATPs), by both facilitated and diffusional mechanisms. This step is competitively inhibited by certain drugs (rifampin, cyclosporine A), which inhibit OATP1B1.
(b) Intracellular Binding
Within the hepatocyte, unconjugated bilirubin binds to cytosolic proteins - primarily glutathione-S-transferases (GSTs), formerly called ligandins, and fatty acid binding proteins. These keep bilirubin in solution and transport it to the endoplasmic reticulum (ER).
(c) Conjugation
In the smooth ER, bilirubin is conjugated with glucuronic acid by bilirubin UDP-glucuronosyltransferase (UGT1A1), encoded by the UGT1A1 gene. Each bilirubin molecule reacts with two UDP-glucuronic acid (UDPGA) molecules to form bilirubin diglucuronide (and monoglucuronide). This conjugation:
- Disrupts internal hydrogen bonds, making bilirubin highly water-soluble
- Is obligatory for biliary excretion
The UGT1 gene complex on chromosome 2 encodes multiple isoforms via alternative first exons (A1-A13), with shared common exons 2-5. Mutations in exon A1 affect only UGT1A1 (bilirubin conjugation), while mutations in common exons (2-5) affect all UGT1 isoforms.
(d) Biliary Excretion
Bilirubin mono- and diglucuronides are excreted across the canalicular membrane into bile via multidrug resistance-associated protein 2 (MRP2, ABCC2), an ATP-dependent active transporter. A portion of glucuronides is also exported back into the portal circulation by MRP3 (ABCC3) on the sinusoidal membrane and undergoes re-uptake by OATP1B1 and OATP1B3 (mutations in these transporters cause Rotor syndrome).
Step 4: Intestinal Metabolism and Enterohepatic Circulation
- Conjugated bilirubin passes down the gut without reabsorption by intestinal mucosa (which is relatively impermeable to it)
- Intestinal bacteria convert bilirubin to a series of colorless compounds called urobilinogens
- Most urobilinogen is excreted in feces (as stercobilin, giving stool its brown color)
- A small fraction is reabsorbed into the portal circulation and undergoes enterohepatic cycling - taken up again by the liver and re-excreted in bile; a small amount escapes hepatic extraction and enters systemic circulation to be excreted in urine as urobilinogen
Renal Excretion
- Unconjugated bilirubin is NOT excreted in urine - too tightly bound to albumin for glomerular filtration; no tubular secretion mechanism
- Conjugated bilirubin (water-soluble) is readily filtered at the glomerulus, hence bilirubinuria occurs in conjugated (direct) hyperbilirubinemia
2. Etiology and Pathogenesis of Jaundice
Jaundice (icterus) is the yellowish discoloration of skin, sclera, and mucous membranes from bilirubin deposition, clinically apparent when serum bilirubin exceeds ~2-3 mg/dL (normal: 0.2-1.0 mg/dL). It results from an imbalance between bilirubin production and hepatobiliary clearance.
Jaundice is classified by mechanism and site of the defect:
A. PRE-HEPATIC (Unconjugated Hyperbilirubinemia - Increased Production)
Mechanism: Excess bilirubin production overwhelms the liver's conjugating capacity.
| Cause | Pathogenesis |
|---|---|
| Hemolysis (sickle cell, G6PD deficiency, hereditary spherocytosis, autoimmune hemolytic anemia, malaria) | Accelerated RBC destruction → increased heme catabolism → increased unconjugated bilirubin. Bilirubin rarely exceeds 4-5 mg/dL with isolated hemolysis (normal liver). Higher values suggest concurrent hepatic dysfunction. Prolonged hemolysis may cause pigment (bilirubin) gallstones. |
| Ineffective erythropoiesis (thalassemia major, megaloblastic anemia, sideroblastic anemia, lead poisoning, porphyria) | Destruction of developing erythroid cells in the marrow before they reach circulation; can account for up to 70% of bilirubin production in severe cases. |
| Resorption of large hematomas / massive tissue infarctions | Degradation of extravasated hemoglobin → transient unconjugated hyperbilirubinemia |
| Massive blood transfusion | Increased fragility of stored erythrocytes → excessive hemoglobin release |
B. HEPATIC (Intrinsic Liver Disease)
Mechanism: Defects in hepatocellular uptake, conjugation, or intrahepatic transport. Can cause mixed or isolated unconjugated/conjugated hyperbilirubinemia.
i. Decreased Hepatic Uptake
- Gilbert syndrome (partial): Reduced uptake contributes to unconjugated hyperbilirubinemia
- Drugs: Rifampin, novobiocin, flavaspidic acid, cyclosporine, cholecystographic contrast agents inhibit OATP1B1 and bilirubin uptake
ii. Impaired Conjugation (Unconjugated Hyperbilirubinemia)
- Physiologic neonatal jaundice: UGT1A1 activity is low at birth; immature intestinal flora lead to enterohepatic circulation of unconjugated bilirubin. Usually resolves by 1-2 weeks.
- Gilbert syndrome: Mild reduction in UGT1A1 activity (due to promoter mutation - insertion in TATA box of UGT1A1 gene); unconjugated bilirubin typically <4 mg/dL; exacerbated by fasting, illness, exercise. Benign.
- Crigler-Najjar Syndrome Type I: Complete absence of UGT1A1 activity; bilirubin 20-45 mg/dL; risk of kernicterus; fatal without treatment (liver transplant).
- Crigler-Najjar Syndrome Type II: Severe reduction (not absence) of UGT1A1; bilirubin 6-25 mg/dL; responds to phenobarbital (induces residual enzyme); kernicterus rare.
- Breast milk jaundice: Prolonged mild unconjugated hyperbilirubinemia in neonates; possibly due to inhibitors of UGT1A1 in breast milk.
iii. Intrahepatic Excretion Defects (Conjugated/Direct Hyperbilirubinemia)
- Dubin-Johnson syndrome: Mutation in MRP2 (ABCC2); impaired canalicular export of conjugated bilirubin. Mild conjugated hyperbilirubinemia, black liver pigment on biopsy, elevated coproporphyrin isomer I in urine. Benign.
- Rotor syndrome: Mutations in both OATP1B1 and OATP1B3; impaired sinusoidal re-uptake of refluxed conjugated bilirubin. Mild conjugated hyperbilirubinemia. Benign.
- Benign recurrent intrahepatic cholestasis (BRIC): Mutations in FIC1 (ATP8B1) or BSEP (ABCB11); recurrent episodes of cholestasis.
- Progressive familial intrahepatic cholestasis (PFIC): Same gene defects as BRIC but more severe; can progress to liver failure.
iv. Hepatocellular Disease (Mixed/Conjugated Hyperbilirubinemia)
- Acute viral hepatitis (HAV, HBV, HCV, HDV, HEV): Hepatocellular necrosis and inflammation → impaired all steps of bilirubin metabolism. Preceded by prodrome of malaise, anorexia, myalgias.
- Drug-induced liver injury (DILI): Acetaminophen (dose-dependent), idiosyncratic drug reactions
- Alcoholic hepatitis: Multifactorial liver injury
- Ischemic hepatitis: Hypotension, hypoxia, hyperthermia, Budd-Chiari syndrome
- Wilson disease: Inherited disorder of copper metabolism; may mimic acute viral hepatitis; hemolytic component adds unconjugated bilirubin
- Chronic liver disease/Cirrhosis: Viral hepatitis (B, C), NAFLD/NASH, alcoholic cirrhosis, hemochromatosis, alpha-1 antitrypsin deficiency, autoimmune hepatitis - jaundice generally signals advanced disease
- Drugs producing intrahepatic cholestasis: Estrogens (down-regulate NTCP, inhibit BSEP and MRP2), anabolic steroids, total parenteral nutrition
- Sepsis-associated cholestasis: Cytokine-mediated down-regulation of NTCP, MRP2, and BSEP
- Paraneoplastic (Stauffer syndrome): Intrahepatic cholestasis without hepatic infiltration, especially renal cell carcinoma and lymphoma; resolves with tumor treatment
C. POST-HEPATIC (Obstructive/Cholestatic) - Conjugated Hyperbilirubinemia
Mechanism: Obstruction to bile flow → backflow of conjugated bilirubin into systemic circulation.
| Cause | Examples |
|---|---|
| Intraluminal obstruction | Choledocholithiasis (common bile duct stones) |
| Mural obstruction | Cholangiocarcinoma, primary sclerosing cholangitis (PSC), biliary strictures, biliary atresia (neonates) |
| Extramural compression | Carcinoma of head of pancreas, ampullary carcinoma, pancreatic pseudocyst, lymph nodes at porta hepatis, Mirizzi syndrome |
| Ampullary pathology | Ampullary carcinoma |
Features of obstructive jaundice: Dark urine (bilirubinuria), pale stools (absent stercobilin), pruritus (bile salt deposition in skin), steatorrhea (fat malabsorption due to absent bile), elevated alkaline phosphatase and GGT predominating over transaminases.
3. Direct vs. Indirect Hyperbilirubinemia
This distinction is based on the van den Bergh reaction (diazo reaction):
- Direct-reacting bilirubin = conjugated bilirubin (water-soluble; reacts directly without alcohol addition)
- Indirect-reacting bilirubin = unconjugated bilirubin (lipid-soluble; requires alcohol addition to react)
- Total bilirubin = direct + indirect
| Feature | Indirect (Unconjugated) Hyperbilirubinemia | Direct (Conjugated) Hyperbilirubinemia |
|---|---|---|
| Chemical form | Free bilirubin (unconjugated) | Bilirubin glucuronide (conjugated) |
| Water solubility | Insoluble (lipid-soluble) | Water-soluble |
| Albumin binding | Tightly bound to albumin | Loosely bound; can dissociate |
| Urine bilirubin (bilirubinuria) | Absent (cannot be filtered) | Present (filtered at glomerulus) |
| Urine color | Normal | Dark ("tea-colored" urine) |
| Stool color | Normal | Pale (acholic stools if obstructive) |
| Site of defect | Pre-hepatic or hepatic (uptake/conjugation defect) | Hepatic (excretion defect) or post-hepatic (obstruction) |
| Blood-brain barrier crossing | YES - lipid-soluble; can cause kernicterus | No |
| Kernicterus risk | Yes (especially neonates) | No |
| Causes | Hemolysis, ineffective erythropoiesis, Gilbert syndrome, Crigler-Najjar, physiologic neonatal jaundice, impaired hepatic uptake (drugs) | Dubin-Johnson, Rotor syndrome, hepatocellular disease (hepatitis, cirrhosis), intrahepatic cholestasis, extrahepatic biliary obstruction |
| Delta bilirubin | Not present | In prolonged conjugated hyperbilirubinemia, bilirubin covalently binds to albumin ("delta bilirubin") - prolonged half-life, not in urine |
| Serum alkaline phosphatase | Normal | Elevated (especially in cholestasis/obstruction) |
| Urine urobilinogen | Elevated (in hemolysis) | Low/absent (in obstruction; no bilirubin reaches gut) |
Key Clinical Distinction
- If direct bilirubin >50% of total: implies hepatocellular disease or biliary obstruction - requires further workup (liver enzymes, imaging)
- If indirect bilirubin predominates: think pre-hepatic (hemolysis) or impaired conjugation
- The direct fraction as measured by clinical labs is typically ≤15% of total in pure hemolysis (Harrison's, Chap. 349)
- Conjugated bilirubin can be filtered and excreted in urine; unconjugated cannot - hence the clinical pearl: "acholuric jaundice" (jaundice without bilirubinuria) = unconjugated hyperbilirubinemia (e.g., hemolysis)
Sources: Harrison's Principles of Internal Medicine 22E, Chap. 349; Sleisenger & Fordtran's Gastrointestinal and Liver Disease; Ganong's Review of Medical Physiology 26E; Schwartz's Principles of Surgery 11E
Describe bilirubin metabolism
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content from four authoritative textbooks with diagrams. Let me compile the full, richly detailed answer.
Bilirubin Metabolism
Bilirubin is a yellow-orange tetrapyrrole pigment and the major end product of heme catabolism. A healthy adult produces approximately 250-350 mg (4 mg/kg) of bilirubin per day. Its metabolism can be divided into five sequential phases: production, plasma transport, hepatic uptake, conjugation, and excretion/enterohepatic circulation.
Phase 1 - Bilirubin Production (Peripheral Tissues)
Sources of Bilirubin
| Source | Contribution |
|---|---|
| Senescent red blood cells (RBC hemoglobin) | 70-85% |
| Ineffective erythropoiesis (intramedullary RBC destruction) | ~15% |
| Non-hemoglobin heme proteins (myoglobin, cytochromes, catalase, peroxidase) | ~15-20% |
RBC hemoglobin degradation occurs primarily in macrophages of the spleen, bone marrow, and hepatic Kupffer cells. Free hemoglobin, haptoglobin-bound hemoglobin, and methemalbuminare catabolized mainly in hepatocytes.
Enzymatic Conversion: Heme → Biliverdin → Bilirubin
The conversion proceeds in two steps:
Step 1 - Heme Oxygenase (HO):
- Opens the porphyrin ring of heme at the α-methene bridge
- Reaction requires NADPH and O₂
- Produces biliverdin (a water-soluble green pigment), releases Fe³+ (recycled for new hemoglobin) and CO (transported as carboxyhemoglobin - a useful marker of hemolytic rate)
- Two isoforms: HO-1 (inducible, ubiquitous, localized to endoplasmic reticulum) and HO-2 (constitutive, expressed selectively in hepatocytes and brain, localized to mitochondria)
Step 2 - Biliverdin Reductase (BVRA):
- Reduces biliverdin IXα to bilirubin IXα (the predominant adult form)
- Reaction requires NADPH (NADPH → NADP⁺)
- BVRA is a ubiquitously expressed cell surface membrane protein
The resulting bilirubin is highly hydrophobic and water-insoluble in its native unconjugated form, due to extensive internal hydrogen bonding that buries the polar groups within the molecule.
Phase 2 - Plasma Transport
Unconjugated (free) bilirubin is tightly and non-covalently bound to albumin in the circulation. This binding:
- Keeps it soluble in plasma
- Prevents its entry into tissues (especially brain) and excretion in urine
- Allows efficient delivery to the liver
Only a very small fraction exists as free (unbound) bilirubin. Certain conditions - hypoalbuminemia, acidosis, competing drugs (sulfonamides, salicylates) - can displace bilirubin from albumin, increasing the free fraction and the risk of kernicterus (especially in neonates).
The half-life of unconjugated bilirubin is very short (~5 minutes); ~60% of labeled bilirubin appears within hepatocytes within 5 minutes of injection.
Phase 3 - Hepatic Uptake
Albumin-bound bilirubin passes through the fenestrated sinusoidal endothelium into the Space of Disse, where it is presented to the basolateral (sinusoidal) membrane of hepatocytes.
Uptake occurs by two mechanisms:
- Passive diffusion - free bilirubin can flip across the membrane
- Carrier-mediated transport - via members of the organic anion transporting polypeptide (OATP) family (particularly OATP1B1 and OATP1B3), which are 12-transmembrane-domain glycoproteins. The exact transporter responsible for unconjugated bilirubin uptake remains debated; competitive inhibition by rifampin and cyclosporine A (which inhibit OATP1B1) provides indirect evidence for a carrier.
This step is highly efficient: clearance of unconjugated bilirubin at normal values is about 5 mg/kg/day.
Inside the hepatocyte, free bilirubin is immediately bound to cytosolic proteins - primarily glutathione-S-transferases (GSTs, formerly called ligandins) (the "Y" and "Z" proteins) and fatty acid binding proteins. These:
- Keep bilirubin in solution (prevent precipitation)
- Prevent its back-diffusion out of the cell
- Shuttle it to the smooth endoplasmic reticulum (SER) by diffusion
Phase 4 - Conjugation
In the smooth endoplasmic reticulum, bilirubin undergoes conjugation with glucuronic acid, catalyzed by bilirubin-UDP-glucuronosyltransferase (UGT1A1):
Bilirubin + 2 UDP-glucuronic acid (UDPGA) → Bilirubin diglucuronide + 2 UDP
Products formed:
- Bilirubin diglucuronide (BDG) - predominant form (~80% in bile)
- Bilirubin monoglucuronide (BMG) - minor form (~20% in bile)
- Small amounts of triglucuronide also form
- Note: The ratio of monoconjugated to diconjugated is ~1:4 in bile, but nearly 1:1 in plasma (monoglucuronides reflux into plasma more readily)
Why conjugation matters:
- Disrupts the internal hydrogen bonds of unconjugated bilirubin
- Converts a lipid-soluble, water-insoluble compound into a highly water-soluble one
- Conjugation is obligatory for excretion into bile
The UGT1 Gene Complex
The UGT1A1 enzyme is encoded by the UGT1A1 gene within the large UGT1 gene complex on chromosome 2. This complex has a unique organization:
- Contains at least 13 substrate-specific first exons (A1, A2 ... A13), each with its own promoter, encoding the amino-terminal ~286 amino acids (substrate-binding domain) of different isoforms
- 4 shared common exons (2-5) encode the carboxyl-terminal half (UDP-glucuronic acid binding site + transmembrane domain)
- Four first exons are pseudogenes; nine functional UGT1 isoforms are expressed
Clinical implication: A mutation in exon A1 affects only UGT1A1 (bilirubin conjugation), while a mutation in common exons 2-5 disrupts ALL UGT1 isoforms (broad substrate conjugation defect, as in Crigler-Najjar Type IA).
Phase 5 - Biliary Excretion
Conjugated bilirubin glucuronides are actively transported against a concentration gradient across the canalicular membrane into the bile canaliculus via:
-
MRP2 (Multidrug Resistance-associated Protein 2, ABCC2) - an ATP-dependent export pump on the canalicular membrane. This is the primary route. Mutations in MRP2 cause Dubin-Johnson syndrome.
-
A portion of glucuronides is also exported back into the portal circulation via MRP3 (ABCC3) on the sinusoidal membrane, then undergoes re-uptake into hepatocytes via OATP1B1 (SLCO1B1) and OATP1B3 (SLCO1B3). Loss-of-function mutations in both OATP1B1 and OATP1B3 cause Rotor syndrome (impaired re-uptake of refluxed conjugated bilirubin).
A small amount of conjugated bilirubin escapes into systemic blood, where it is loosely bound to albumin (less tightly than unconjugated bilirubin) and can be filtered at the glomerulus and excreted in urine as bilirubinuria.
Phase 6 - Intestinal Metabolism and Enterohepatic Circulation
After secretion into bile, conjugated bilirubin passes through bile ducts → enters the duodenum → travels down the gastrointestinal tract.
Key points:
- The intestinal mucosa is relatively impermeable to conjugated bilirubin - it is NOT absorbed in the small intestine
- In the colon, intestinal bacteria deconjugate and enzymatically reduce bilirubin to a series of colorless compounds collectively called urobilinogens (including mesobilinogen, stercobilinogen)
- Most urobilinogen (~80%) is oxidized and excreted in feces as stercobilin - this gives stool its characteristic brown color
Enterohepatic Circulation of Urobilinogen
- A fraction of urobilinogen (~20%) is reabsorbed from the colon into the portal circulation
- The liver re-extracts and re-excretes the majority back into bile (enterohepatic circulation)
- A small amount escapes hepatic extraction, enters systemic circulation, and is filtered by the kidneys and excreted in urine as urobilinogen
Note: Unconjugated bilirubin does NOT normally reach the gut in adults. It only does so in neonates or in severe unconjugated hyperbilirubinemia (e.g., Crigler-Najjar Type I). If it reaches the gut, it can be partly reabsorbed, amplifying the hyperbilirubinemia.
Renal Excretion - Summary
| Form | Albumin Binding | Glomerular Filtration | Urine |
|---|---|---|---|
| Unconjugated bilirubin | Tight | NOT filtered | Absent (acholuric) |
| Conjugated bilirubin | Loose | Readily filtered | Present (bilirubinuria) |
| Urobilinogen | Minimal | Filtered | Small amount normally present |
Delta Bilirubin (δ-Bilirubin) - Clinically Important
In prolonged conjugated hyperbilirubinemia, a proportion of conjugated bilirubin covalently bonds to albumin to form delta bilirubin (δ-bilirubin). This has the same half-life as albumin (~3 weeks), which explains why jaundice and elevated serum bilirubin persist for weeks even after successful resolution of the underlying liver disease or biliary obstruction. Delta bilirubin is measured in the direct bilirubin fraction but is NOT excreted in urine (being albumin-bound).
Summary Flowchart
HEME (from senescent RBCs, hemoproteins)
↓ Heme oxygenase (NADPH + O₂ → CO + Fe³⁺ + NADP⁺)
BILIVERDIN (water-soluble, green)
↓ Biliverdin reductase (NADPH → NADP⁺)
UNCONJUGATED BILIRUBIN (lipid-soluble, yellow-orange)
↓ Bound to albumin in plasma
LIVER HEPATOCYTE
↓ Uptake via OATP/diffusion; bound to GST (ligandin)
SMOOTH ER → UGT1A1 conjugation with UDP-glucuronic acid
↓
CONJUGATED BILIRUBIN (water-soluble diglucuronide)
↓ MRP2 (ATP-dependent) → Bile canaliculus
BILE DUCTS → DUODENUM → INTESTINE
↓ Bacterial action
UROBILINOGEN
↙ ↘
Excreted in feces Reabsorbed → Portal vein
(stercobilin-brown) ↙ ↘
Re-excreted Urine urobilinogen
in bile
Normal Serum Bilirubin Values
| Fraction | Normal range |
|---|---|
| Total bilirubin | 0.2 - 1.0 mg/dL (up to 1.2 mg/dL) |
| Direct (conjugated) | 0 - 0.3 mg/dL |
| Indirect (unconjugated) | 0.2 - 0.8 mg/dL |
Jaundice becomes clinically visible when total bilirubin exceeds ~2-3 mg/dL.
Sources: Harrison's Principles of Internal Medicine 22E (Chap. 349); Ganong's Review of Medical Physiology 26E (Chap. 28); Henry's Clinical Diagnosis and Management by Laboratory Methods (Chap. 22); Yamada's Textbook of Gastroenterology 7E
Enumerate the etiology and pathogenesis of jaundice
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
I now have comprehensive content from multiple authoritative sources. Let me compile the full, well-organized answer.
Etiology and Pathogenesis of Jaundice
Jaundice (icterus) is the yellowish discoloration of the skin, sclera, and mucous membranes due to deposition of bilirubin pigment. It becomes clinically visible when serum bilirubin exceeds ~2.5-3 mg/dL (normal: 0.2-1.0 mg/dL).
The fundamental mechanism is always the same: an imbalance between bilirubin production and its hepatobiliary clearance. Jaundice is best classified by the anatomical phase of bilirubin metabolism that is disrupted.
Classification Framework
JAUNDICE
│
├── I. PRE-HEPATIC
│ └── Increased bilirubin production
│ → Unconjugated (indirect) hyperbilirubinemia
│
├── II. HEPATIC (Intra-hepatic)
│ ├── A. Decreased hepatic uptake
│ ├── B. Impaired conjugation → Unconjugated hyperbilirubinemia
│ ├── C. Impaired canalicular excretion
│ └── D. Hepatocellular disease → Mixed/conjugated hyperbilirubinemia
│
└── III. POST-HEPATIC
└── Biliary obstruction → Conjugated (direct) hyperbilirubinemia
I. PRE-HEPATIC JAUNDICE
Mechanism: Bilirubin is produced at a rate that exceeds the liver's capacity to conjugate and excrete it. The result is unconjugated (indirect) hyperbilirubinemia.
Key point: Even with complete hemolysis, the bone marrow can sustain at most an 8-fold increase in RBC production. Thus, hemolysis alone cannot sustain serum bilirubin above ~4 mg/dL in a liver with normal function. Higher levels imply concurrent hepatic dysfunction.
1. Hemolysis (Most Common Pre-hepatic Cause)
Etiology:
| Type | Examples |
|---|---|
| Inherited hemolytic anemias | Sickle cell disease, hereditary spherocytosis, G6PD deficiency, thalassemia |
| Immune-mediated (acquired) | Autoimmune hemolytic anemia (warm/cold type), transfusion reactions, drug-induced (methyldopa, penicillin) - Coombs test positive |
| Non-immune acquired | Microangiopathic hemolytic anemia (TTP, HUS, DIC), mechanical hemolysis (prosthetic heart valves), infections (malaria, Clostridium perfringens), drugs/toxins directly damaging RBCs |
Pathogenesis:
- Accelerated RBC destruction → massive release of hemoglobin → heme catabolism exceeds hepatic conjugating capacity → accumulation of unconjugated bilirubin
- Bilirubin remains unconjugated; direct fraction ≤15% of total
- Prolonged hemolysis → precipitation of bilirubin salts in the biliary tree → pigment (bilirubin) gallstones → secondary obstructive jaundice
2. Ineffective Erythropoiesis
Etiology: Thalassemia major, megaloblastic anemias (folate/B12 deficiency), sideroblastic anemia, congenital erythropoietic porphyria, lead poisoning, dyserythropoietic anemias
Pathogenesis:
- Developing erythroid cells are destroyed within the bone marrow before entering circulation
- Normally only a small fraction of bilirubin comes from intramedullary destruction; in these disorders it can reach up to 70% of total bilirubin production
- Results in modest unconjugated hyperbilirubinemia
3. Resorption of Hematoma / Tissue Infarction
Pathogenesis: Extravascular hemoglobin in large hematomas or massive tissue infarctions undergoes local catabolism → transient unconjugated hyperbilirubinemia
4. Massive Blood Transfusion
Pathogenesis: Stored RBCs have increased fragility → excessive hemoglobin release → overwhelms hepatic conjugation
5. Hypoalbuminemia (Defective Transport)
Pathogenesis: Severe protein deficiency (burns, nephrotic syndrome, malnutrition) → inadequate albumin for bilirubin transport → reduced delivery to liver and impaired uptake
II. HEPATIC (INTRAHEPATIC) JAUNDICE
Defects at the level of the hepatocyte - in uptake, conjugation, or canalicular excretion.
A. Decreased Hepatic Uptake → Unconjugated Hyperbilirubinemia
Etiology and Pathogenesis:
| Cause | Mechanism |
|---|---|
| Gilbert syndrome (partial) | Reduced OATP-mediated sinusoidal uptake contributes alongside impaired conjugation |
| Drugs: rifampin, novobiocin, flavaspidic acid, cyclosporine A, cholecystographic contrast agents | Competitive inhibition of OATP1B1 on the sinusoidal membrane → impaired uptake of unconjugated bilirubin. Resolves on stopping the drug. |
| Prolonged fasting | Reduces hepatic uptake capacity |
B. Impaired Conjugation → Unconjugated Hyperbilirubinemia
(i) Physiologic Neonatal Jaundice
Pathogenesis:
- Fetal bilirubin was cleared by the placenta and maternal liver
- At birth, the neonatal liver suddenly assumes responsibility for clearance
- UGT1A1 activity is low at birth (immature)
- Intestinal flora that convert bilirubin to urobilinogen are also undeveloped
- Alternative excretory pathways allow passage of unconjugated bilirubin into the gut, where it is reabsorbed (enterohepatic circulation of unconjugated bilirubin)
- Result: Nearly all neonates develop mild unconjugated hyperbilirubinemia in the first week of life; typically resolves by 1-2 weeks
(ii) Gilbert Syndrome
Etiology: Promoter mutation in the UGT1A1 gene - a TA insertion in the TATA box (A(TA)₇TAA instead of normal A(TA)₆TAA) → reduced UGT1A1 transcription → ~30% reduction in enzyme activity. Autosomal recessive inheritance. Prevalence: 6-12% of the population.
Pathogenesis:
- Mild reduction in bilirubin conjugation + decreased hepatic uptake
- Serum bilirubin typically <4 mg/dL (usually unconjugated)
- Exacerbated by fasting, physical stress, illness, intercurrent infections, alcohol
- Entirely benign; no liver damage
(iii) Crigler-Najjar Syndrome, Type I (CN-I)
Etiology: Autosomal recessive; mutations in UGT1A1 gene - complete absence of functional UGT1A1 activity. Prevalence: 0.6-1.0 per million. Type IA: mutations in common exons 2-5 (affects all UGT1 substrates). Type IB: mutations in exon A1 (bilirubin-specific).
Pathogenesis:
- Total absence of bilirubin conjugation
- Bilirubin diglucuronide is virtually absent from bile
- Serum unconjugated bilirubin: 20-45 mg/dL (340-765 µmol/L)
- Bilirubin eliminated only by alternative pathways: direct passage into bile and small intestine as bilirubin photoisomers
- No response to phenobarbital (no residual enzyme to induce)
- Unconjugated bilirubin crosses the blood-brain barrier → kernicterus (bilirubin encephalopathy) - usual outcome without treatment
- Fatal without liver transplantation
(iv) Crigler-Najjar Syndrome, Type II (CN-II)
Etiology: Autosomal recessive; missense mutations in UGT1A1 gene - severely reduced (not absent) UGT1A1 activity.
Pathogenesis:
- Serum unconjugated bilirubin: 6-25 mg/dL (usually <20 mg/dL)
- Small amount of bilirubin glucuronide present in bile (predominantly monoglucuronide)
- Responds to phenobarbital (induces residual enzyme) - serum bilirubin falls >25%
- Kernicterus rare (occurs mainly with physiologic stress, illness, fasting)
- Long-term prognosis better than CN-I; some patients managed with phenobarbital + phototherapy
(v) Breast Milk Jaundice
Pathogenesis: Certain fatty acids in breast milk inhibit UGT1A1 activity; increased enterohepatic circulation of bilirubin may also contribute. Distinct from Lucey-Driscoll syndrome (transient familial neonatal hyperbilirubinemia), where a UGT1A1 inhibitor is present in maternal serum.
(vi) Acquired Conjugation Defects
Pathogenesis: Advanced hepatitis or cirrhosis → modest reduction in UGT1A1 activity (though conjugation is better preserved than canalicular excretion in hepatocellular disease). Drugs - pregnanediol, chloramphenicol, gentamicin, atazanavir - inhibit UGT1A1 activity.
C. Impaired Canalicular Excretion of Conjugated Bilirubin → Conjugated Hyperbilirubinemia
(i) Dubin-Johnson Syndrome (DJS)
Etiology: Autosomal recessive; mutations in MRP2 (ABCC2) gene - the ATP-dependent canalicular export pump for conjugated bilirubin and organic anions.
Pathogenesis:
- Conjugated bilirubin formed normally but cannot be exported into the bile canaliculus
- Refluxes back into plasma → conjugated hyperbilirubinemia + bilirubinuria
- Serum bilirubin typically 2-5 mg/dL (predominantly conjugated); occasionally higher
- Liver appears grossly black due to accumulation of dark melanin-like pigment in hepatocytes (lysosomal)
- Diagnostic hallmark: urinary coproporphyrin isomer I ≥80% of total (normally ~25%)
- Otherwise completely benign; normal liver function tests (ALP, transaminases); normal life expectancy
(ii) Rotor Syndrome (RS)
Etiology: Autosomal recessive; simultaneous loss-of-function mutations in both OATP1B1 (SLCO1B1) and OATP1B3 (SLCO1B3).
Pathogenesis:
- These transporters normally mediate sinusoidal re-uptake of conjugated bilirubin that has refluxed from the hepatocyte into the portal circulation
- Loss of both → conjugated bilirubin cannot be recaptured → accumulates in plasma
- Mild conjugated hyperbilirubinemia; bilirubinuria present
- Liver has no abnormal pigmentation (unlike DJS)
- Total urinary coproporphyrin is substantially increased (distinguishes from DJS); gallbladder usually visualized on oral cholecystography (opposite of DJS)
- Benign, normal life expectancy
(iii) Benign Recurrent Intrahepatic Cholestasis (BRIC) and Progressive Familial Intrahepatic Cholestasis (PFIC)
| Feature | PFIC1/BRIC1 | PFIC2/BRIC2 | PFIC3 |
|---|---|---|---|
| Gene | ATP8B1 (FIC1) | ABCB11 (BSEP) | ABCB4 (MDR3) |
| Protein | FIC1 (phospholipid flippase) | BSEP (bile salt export pump) | MDR3 (phospholipid transporter) |
| GGT | Normal | Normal | Markedly elevated |
| Pathogenesis | FIC1 dysfunction → ↑susceptibility to bile acid damage, ↓BSEP expression | Impaired bile salt secretion → ↓bile flow → cholestasis | ↓phospholipid in bile → bile acid-induced cholangiopathy |
BRIC: Episodic (recurrent) cholestasis; does NOT progress to liver failure.
PFIC: Progressive; can lead to cirrhosis and liver failure in childhood.
D. Hepatocellular Disease → Mixed (Unconjugated + Conjugated) Hyperbilirubinemia
When hepatocytes are diffusely injured, multiple steps of bilirubin metabolism fail simultaneously (uptake, conjugation, and canalicular excretion), producing a mixed picture.
(i) Acute Hepatocellular Disease
| Cause | Pathogenesis |
|---|---|
| Acute viral hepatitis (HAV, HBV, HCV, HDV, HEV) | Hepatocellular necrosis and inflammation → failure of uptake, conjugation, and excretion. HAV and HEV are enterally transmitted, self-limited. HBV, HCV, HDV are parenterally transmitted and may become chronic. Prodrome of malaise, anorexia, myalgias precedes jaundice. |
| Drug-induced liver injury (DILI) | Acetaminophen (dose-dependent, zone 3 necrosis via NAPQI); idiosyncratic drug reactions (amoxicillin-clavulanate, isoniazid, etc.) → hepatocellular necrosis |
| Alcoholic hepatitis | Toxic metabolites of alcohol (acetaldehyde) → hepatocellular injury + Mallory-Denk bodies; cytokine-driven inflammation |
| Ischemic hepatitis ("shock liver") | Hypoperfusion, hypoxia, hyperthermia → centrilobular necrosis; markedly elevated transaminases; causes: hypotension, cardiac failure, Budd-Chiari syndrome, sinusoidal obstruction syndrome |
| Wilson disease | Copper accumulation → hepatocellular injury mimicking acute viral hepatitis; hemolytic component adds unconjugated bilirubin; diagnosed by slit-lamp (Kayser-Fleischer rings), low serum ceruloplasmin |
| Sepsis-associated jaundice | Inflammatory cytokines (TNF-α, IL-6) → rapid, reversible down-regulation of MRP2, NTCP, and BSEP transporters → decreased canalicular excretion → conjugated hyperbilirubinemia (usually <5 mg/dL); resolves with treatment of infection |
(ii) Intrahepatic Cholestasis (Drug/Hormone-Induced)
| Cause | Pathogenesis |
|---|---|
| Oral contraceptives / estrogens | Down-regulate NTCP (bile salt uptake) and competitively inhibit BSEP and MRP2 → impaired bile salt secretion and canalicular transport. Develops within 2 months; resolves on stopping. |
| Anabolic steroids | Same mechanism as estrogens → bland intrahepatic cholestasis |
| Total parenteral nutrition (TPN) | Altered enterohepatic circulation; diminished neuroendocrine stimulation of bile flow → cholestasis; may progress to fibrosis |
(iii) Chronic Hepatocellular Disease / Cirrhosis
Jaundice in chronic liver disease typically signals advanced disease (cirrhosis). Causes include:
- Chronic viral hepatitis B and C
- Alcoholic cirrhosis (ARLD)
- Non-alcoholic fatty liver disease (NAFLD/NASH) → NASH cirrhosis
- Hemochromatosis (HFE gene mutations; decades of iron overload)
- Alpha-1-antitrypsin deficiency (misfolded ZZ-phenotype protein accumulates in hepatocyte ER)
- Wilson disease (copper accumulation → progressive fibrosis)
- Autoimmune hepatitis
(iv) Paraneoplastic (Stauffer Syndrome)
Pathogenesis: Jaundice in absence of hepatic infiltration by tumor; most classically with renal cell carcinoma and lymphoma. Tumor-derived cytokines down-regulate NTCP, MRP2, and BSEP. Resolves with successful tumor treatment.
III. POST-HEPATIC (OBSTRUCTIVE / CHOLESTATIC) JAUNDICE
Mechanism: Obstruction to bile flow at any level → conjugated bilirubin accumulates in hepatocytes → refluxes back into the sinusoidal blood → conjugated (direct) hyperbilirubinemia + bilirubinuria + pale stools + pruritus.
Intraluminal Obstruction (Within the Bile Duct Lumen)
- Choledocholithiasis (common bile duct stones) - most common cause; stone may originate from gallbladder or form de novo in the duct; often complicates prolonged hemolysis (pigment stones)
- Biliary parasites (Clonorchis sinensis, Ascaris lumbricoides)
- Blood clots (hemobilia)
Mural Obstruction (Wall of Bile Duct)
- Cholangiocarcinoma (Klatskin tumor at the hilum; intrahepatic; distal CBD)
- Primary sclerosing cholangitis (PSC) - autoimmune fibro-inflammatory stricturing of intra- and extrahepatic bile ducts; strongly associated with ulcerative colitis
- Benign biliary strictures - post-operative (surgical clips, ischemic injury), post-inflammatory
- Cholangitis (acute bacterial cholangitis - infection superimposed on obstruction)
- Biliary atresia (neonates - fibro-obliterative process of bile ducts; presents as neonatal cholestatic jaundice)
- Caroli disease (congenital dilation of intrahepatic bile ducts)
Extramural Compression (External Compression of Bile Duct)
- Carcinoma of head of pancreas - most common malignant cause of obstructive jaundice; compresses distal CBD → painless progressive jaundice (Courvoisier's sign)
- Ampullary carcinoma - jaundice + often GI bleeding
- Chronic pancreatitis - fibrotic stricture of the intrapancreatic CBD
- Pancreatic pseudocyst - external compression
- Mirizzi syndrome - large gallstone in the cystic duct compresses the common hepatic duct
- Lymphadenopathy at the porta hepatis - metastatic nodes (gastric, colorectal, breast cancer)
- Retroperitoneal fibrosis
Surgical Complications
- Misplaced surgical clips, retained stones, inadvertent ischemic injury to biliary system during cholecystectomy or hepatobiliary surgery → obstructive jaundice (may present immediately or years later)
Summary Table: Key Features by Mechanism
| Feature | Pre-hepatic | Hepatocellular | Obstructive |
|---|---|---|---|
| Bilirubin type elevated | Indirect (unconjugated) | Both (mixed) | Direct (conjugated) |
| Urine color | Normal | Dark | Dark |
| Stool color | Normal | Normal/pale | Pale (acholic) |
| Bilirubinuria | Absent | Present | Present |
| Urine urobilinogen | Increased | Variable | Absent/decreased |
| Pruritus | Absent | Variable | Present (prominent) |
| Alkaline phosphatase | Normal | Mildly elevated | Markedly elevated |
| ALT/AST | Normal | Markedly elevated | Mildly elevated |
| Blood film / Coombs | Abnormal (hemolysis) | Normal | Normal |
Sources: Harrison's Principles of Internal Medicine 22E (Chap. 349); Sleisenger & Fordtran's Gastrointestinal and Liver Disease; Schwartz's Principles of Surgery 11E (Chap. 30); Yamada's Textbook of Gastroenterology 7E
This is a shared conversation. Sign in to Orris to start your own chat.