Enlighten me about beta arrestin pathway that is responsible for respiratory depression in morphine administration
Reading File
Finding Sources
Finding Sources
Searching PubMed
beta-arrestin opioid respiratory depression morphine
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Reading File
Loading Image
Finding Sources
Searching the Web
beta-arrestin2 morphine respiratory depression controversy oliceridine 2023 2024
I now have comprehensive, rich content from authoritative textbooks and the latest web evidence including the Kliewer et al. controversy paper. I have everything needed for a thorough, well-structured answer.
Beta-Arrestin Pathway and Morphine-Induced Respiratory Depression
1. The mu-Opioid Receptor (MOR) and Dual Signaling
Morphine binds the mu-opioid receptor (MOR), a class A G-protein-coupled receptor (GPCR). When activated, MOR can signal through two major intracellular routes simultaneously, not sequentially:
- G-protein pathway (Gi/o) - responsible for analgesia
- Beta-arrestin pathway - historically attributed to adverse effects including respiratory depression, constipation, tolerance, and euphoria
The diagram below from Goodman & Gilman shows the full sequence of MOR activation and the role of each pathway:
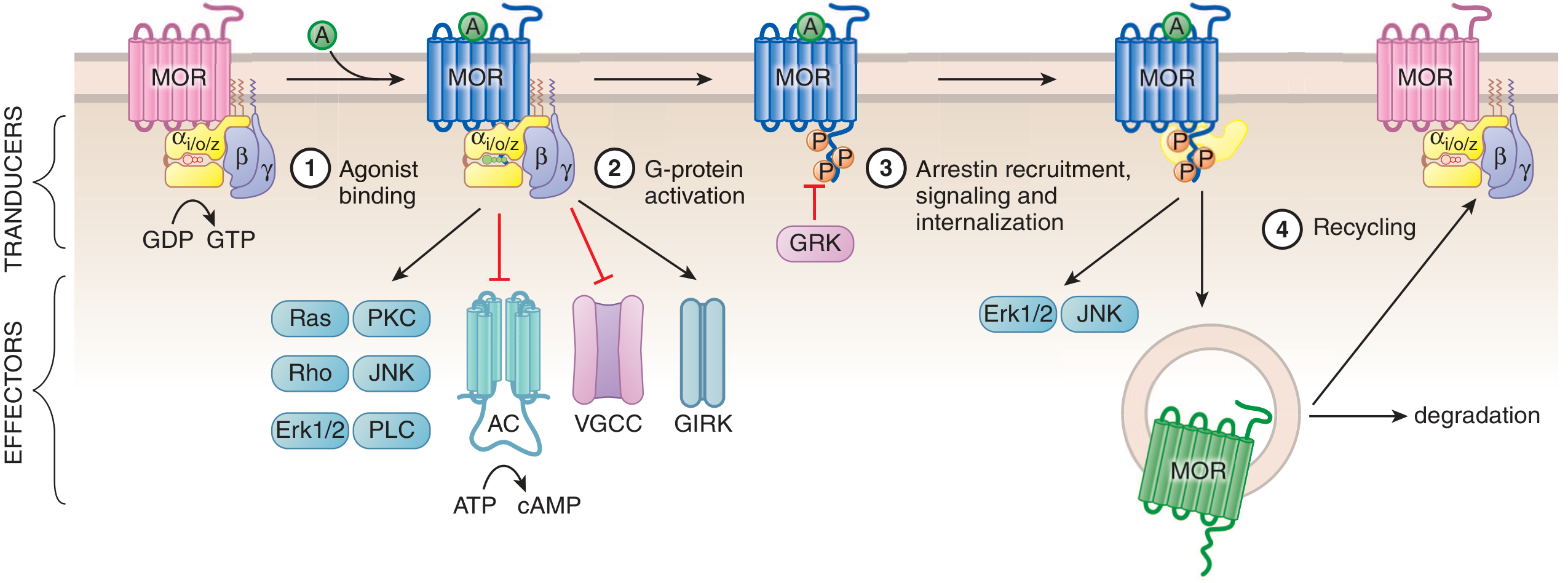
Steps: (1) Agonist binding converts inactive MOR (Ri) to active MOR (Ra). (2) G-protein activation with GDP→GTP exchange, leading to inhibition of adenylyl cyclase (AC), voltage-gated Ca²⁺ channels (VGCC), and opening of GIRK K⁺ channels. (3) GRK phosphorylates the receptor, recruiting beta-arrestin for internalization and independent scaffolding signaling. (4) Internalized receptor is either degraded or recycled.
2. Step-by-Step: The Beta-Arrestin Signaling Cascade
Step 1 - Agonist Binding and G-protein Activation
Morphine binds the orthosteric site on MOR, causing the alpha subunit of the Gi/o heterotrimer to exchange GDP for GTP. The activated alpha subunit directly inhibits adenylyl cyclase, reducing cAMP. The beta-gamma dimer:
- Inhibits voltage-gated Ca²⁺ channels (reduces neurotransmitter release)
- Opens GIRK K⁺ channels (hyperpolarization)
- Activates MAPK cascades
- Recruits GRK2 and GRK3 to the receptor
Step 2 - GRK-Mediated Receptor Phosphorylation
The beta-gamma dimer recruits G-protein receptor kinases (GRK2 and GRK3) to phosphorylate the cytoplasmic tail and intracellular loops of MOR. This phosphorylation is the critical prerequisite for beta-arrestin recruitment. PKC can also phosphorylate the receptor independently of GRK.
Step 3 - Beta-Arrestin Recruitment and Scaffolding
Phosphorylated MOR recruits beta-arrestin 2 (beta-arr2). This recruitment has two major consequences:
- Steric uncoupling from G-protein: Beta-arrestin physically displaces Gi/o from the receptor's cytoplasmic core, desensitizing the G-protein pathway ("homologous desensitization")
- Independent scaffolding signaling: Beta-arrestin acts as a scaffold for the MAPK/ERK cascade, Src kinase, and Akt/PI3K pathways - generating its own signaling outputs distinct from G-protein signaling
Step 4 - Receptor Internalization
The beta-arrestin-receptor complex undergoes clathrin-mediated endocytosis. Within endosomes, the receptor-beta-arrestin complex can continue signaling ("endosomal signaling"). The receptor is then either sent to lysosomes for degradation or dephosphorylated and recycled to the plasma membrane.
The diagram below from Miller's Anesthesia illustrates the full beta-arrestin 2 lifecycle including the downstream MAPK, Src, and Akt signaling:
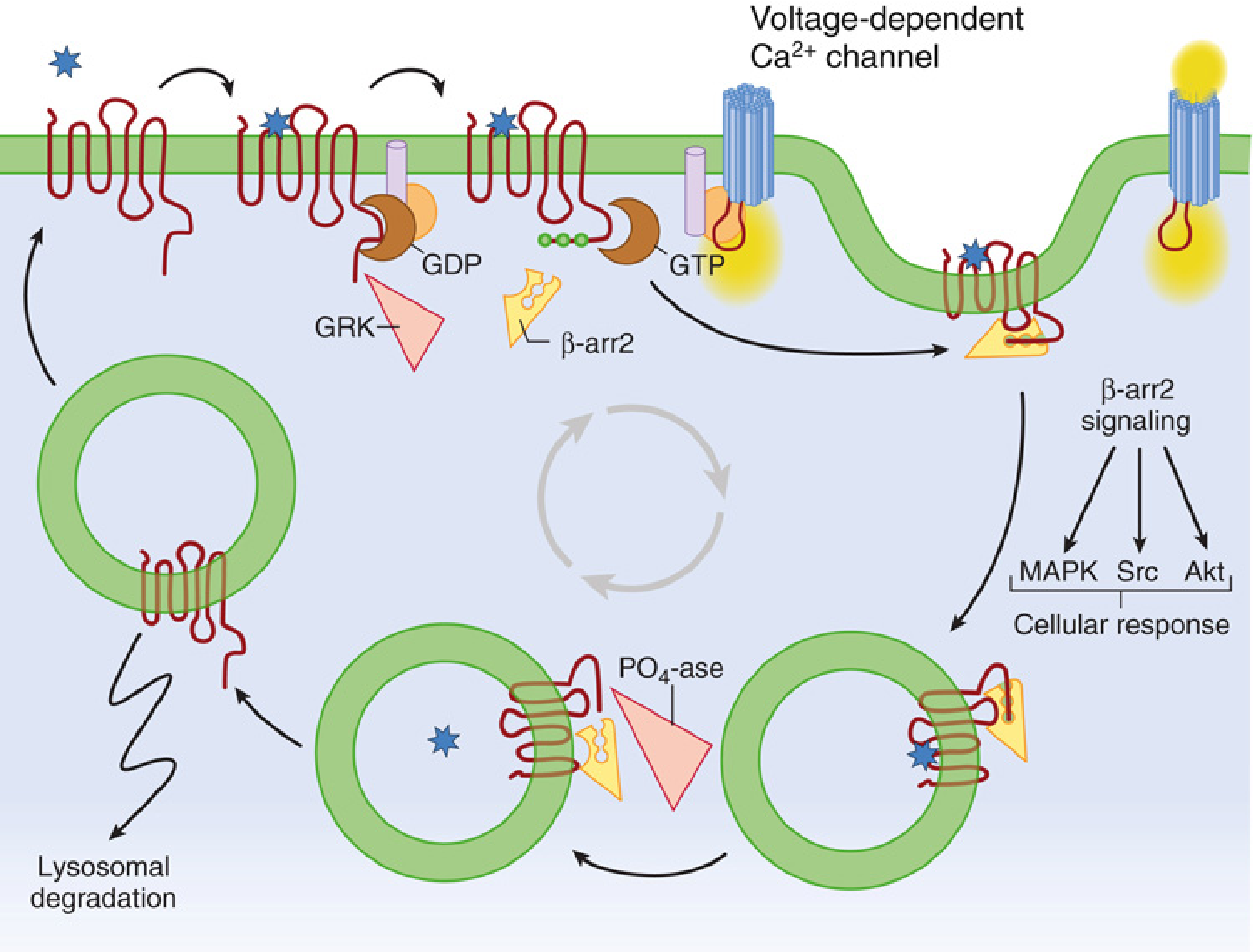
3. How Beta-Arrestin Causes Respiratory Depression
The respiratory depression from morphine is mediated in pre-Botzinger complex neurons and other brainstem respiratory control centers. The proposed mechanism is:
- Beta-arrestin 2 scaffolds ERK1/2 in brainstem respiratory neurons, altering neuronal firing patterns
- Beta-arrestin-mediated receptor desensitization and downregulation reduces the receptor density over time, necessitating higher doses for equivalent analgesia - progressively increasing respiratory depression risk
- Beta-arrestin signaling activates Src kinase, which can modulate voltage-gated potassium channels in respiratory neurons, suppressing their firing rate
- The endosomal MAPK signaling complex (receptor + beta-arrestin + ERK1/2) may have prolonged and spatially distinct effects compared to G-protein signaling at the plasma membrane
In contrast, the G-protein (Gi/o) pathway mediates analgesia primarily through:
- cAMP reduction in spinal cord dorsal horn neurons
- GIRK channel activation causing hyperpolarization
- Inhibition of VGCC reducing transmitter release at primary afferents
As stated in Miller's Anesthesia: "The Gi/o signaling pathway was thought to mediate analgesic action of morphine, whereas beta-arrestin signaling results in unwanted side effects including euphoria, respiratory depression, and gastrointestinal effects." - Miller's Anesthesia, 10e
4. Biased Agonism - The Concept of Separating Analgesia from Adverse Effects
The dissociation of these two pathways gave rise to the concept of "biased agonism" (also called functional selectivity). A G-protein-biased agonist would preferentially activate the Gi/o analgesic pathway while minimizing beta-arrestin recruitment, theoretically delivering analgesia without respiratory depression.
Key supporting evidence for the beta-arrestin hypothesis included:
- Beta-arrestin 2 knockout mice (Raehal et al., 2005): Morphine's analgesic properties were enhanced and respiratory depression + constipation were attenuated in these animals - the foundational study for the biased agonism concept
- TRV130 (oliceridine): Designed as a G-protein-biased MOR agonist with markedly reduced receptor phosphorylation and beta-arrestin 2 recruitment. In a randomized, double-blind, placebo-controlled crossover study in healthy volunteers, TRV130 produced greater analgesia than morphine with less respiratory depression and less severe nausea
- PZM21: Identified computationally as a highly G-protein-biased compound that suppressed pain in mice without constipation, respiratory depression, hyperlocomotion, or addiction-related behaviors
5. The Controversy: Is Beta-Arrestin Really the Culprit?
The original beta-arrestin hypothesis has been substantially challenged by more recent evidence:
Miller's Anesthesia notes: "Contrary to the previous report, it was recently reported that the morphine-induced respiratory depression in beta-arrestin 2-deficient mice was not significantly different from that observed in wild-type mice."
Kliewer et al. (2022) - a multi-laboratory consortium study - found that morphine (3.75-100 mg/kg) and fentanyl induced respiratory depression and constipation in beta-arrestin 2 knockout mice indistinguishably from wild-type mice. Their conclusion was unequivocal: beta-arrestin 2 signaling does not play a key role in opioid-induced respiratory depression. This paper calls into question whether developing G-protein-biased agonists is the right strategy for safer opioids.
Proposed alternative explanations include:
- The apparent safety of TRV130 may reflect its lower intrinsic efficacy (partial agonism) rather than G-protein bias per se
- Its unique pharmacokinetic profile (faster offset) may explain the seemingly reduced adverse effects
- Respiratory depression may be mediated by other signaling molecules - possibly GRK3, PKC, or alternative arrestin isoforms (beta-arrestin 1)
- Endogenous opioids like enkephalins, which are relatively "balanced" agonists, may paradoxically show less adverse effect profile than strongly G-protein-biased synthetic ligands - as noted in Kaplan & Sadock: "truly balanced opioids similar to endogenous enkephalins may be more beneficial in limiting opioid dependence"
6. Clinical Translation - Oliceridine (TRV130)
Despite the mechanistic controversy, oliceridine was FDA-approved in August 2020 for moderate-to-severe acute postoperative pain. Key clinical points (Barash's Clinical Anesthesia, 9e):
| Parameter | Detail |
|---|---|
| Mechanism | G-protein-biased MOR agonist |
| Equianalgesic dose | 1 mg IV oliceridine ≈ 5 mg IV morphine |
| Dosing | 1-2 mg IV q1-3h PRN; max 27 mg/24h |
| Metabolism | CYP3A4 + CYP2D6 (hepatic) |
| Adverse effects | Dose-dependent respiratory depression still reported; reward-enhancing effects similar to morphine/oxycodone |
| Limitation | Abuse potential not eliminated |
Summary Table
| Feature | G-Protein (Gi/o) Pathway | Beta-Arrestin Pathway |
|---|---|---|
| Trigger | MOR agonist binding | GRK phosphorylation of MOR |
| Mediators | Alpha subunit (AC inhibition), beta-gamma (VGCC, GIRK, MAPK) | Beta-arr2 recruitment, MAPK/ERK, Src, Akt |
| Primary effect | Analgesia, sedation | Respiratory depression, constipation, tolerance, dependence (classic view) |
| Receptor fate | Receptor remains at membrane | Endocytosis, degradation or recycling |
| Drug exploiting bias | Oliceridine (TRV130) - G-protein biased | Morphine (balanced agonist) |
| Knockout mouse data | - | Contradictory (Raehal 2005 vs. Kliewer 2022) |
Bottom line: The beta-arrestin 2 pathway, activated downstream of GRK-mediated MOR phosphorylation, was the dominant mechanistic explanation for morphine-induced respiratory depression for over a decade, driving development of G-protein-biased agonists like oliceridine. However, recent multi-laboratory knockout studies have challenged this simple model. The current consensus is that the picture is more nuanced - beta-arrestin signaling likely contributes to tolerance and constipation, while respiratory depression may involve additional mechanisms. The field is actively re-evaluating the biased agonism paradigm.
Sources: Goodman & Gilman's Pharmacological Basis of Therapeutics, 13e | Miller's Anesthesia, 10e | Barash's Clinical Anesthesia, 9e | Kaplan & Sadock's Comprehensive Textbook of Psychiatry | Kliewer et al., 2022 (PMID: referenced in Miller's)
This is a shared conversation. Sign in to Orris to start your own chat.