Tell me anti histamine drugs from pharmacology from basics to clinical
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Excellent - I have a wealth of content from Goodman & Gilman's. Now I have everything needed to write a comprehensive answer. Let me compile the full response.
Antihistamine Drugs: Basics to Clinical
Based primarily on Goodman & Gilman's The Pharmacological Basis of Therapeutics, supplemented by Stahl's Essential Psychopharmacology and Miller's Anesthesia.
1. Histamine - The Foundation
Before understanding antihistamines, you need to understand histamine itself.
Synthesis and Storage
- Histamine is formed by decarboxylation of L-histidine (enzyme: histidine decarboxylase)
- Stored in mast cell granules and basophil granules bound to heparin proteoglycans
- Also found in enterochromaffin-like (ECL) cells of the gastric mucosa and histaminergic neurons of the CNS (tuberoammillary nucleus of the posterior hypothalamus)
Histamine Receptors
| Receptor | Location | Second Messenger | Key Effect |
|---|---|---|---|
| H1 | Smooth muscle, endothelium, CNS neurons, nerve endings | Gq → PLC → IP3/DAG → ↑Ca²⁺ | Bronchoconstriction, itch, vasodilation via NO, allergic responses |
| H2 | Gastric parietal cells, heart, smooth muscle | Gs → ↑cAMP | Gastric acid secretion, cardiac stimulation, vasodilation |
| H3 | Presynaptic CNS (autoreceptor), peripheral nerves | Gi → ↓cAMP | Feedback inhibition of histamine synthesis/release; modulates other NT release |
| H4 | Hematopoietic cells, mast cells, leukocytes | Gi → ↓cAMP | Chemotaxis, cytokine secretion, immune modulation |
Pathophysiological Actions of Histamine
Triple Response of Lewis (intradermal injection):
- Initial red spot - direct H1-mediated vasodilation (NO production)
- Flare - axonal reflex-mediated indirect vasodilation
- Wheal - ↑capillary permeability → edema
Cardiovascular: Vasodilation → ↓BP; ↑heart rate (H2); AV conduction slowing (H1); ↑contractility (H2)
Respiratory: Bronchoconstriction (H1 dominant in humans) - asthmatics are hypersensitive
GI: Gastric acid secretion via H2 on parietal cells
CNS: Promotes wakefulness, cognition, locomotion (stimulatory); suppresses appetite and convulsions (inhibitory)
Nerve endings: H1 mediates itch in epidermis; pain in dermis
2. Classification of Antihistamines
Important pharmacological note: H1 antagonists are technically inverse agonists - they reduce constitutive receptor activity AND compete with histamine. They don't just block; they reverse baseline receptor tone.
H1 Antihistamines (Anti-allergic)
Generation 1 vs Generation 2 - Key Comparison
| Property | 1st Generation | 2nd Generation |
|---|---|---|
| CNS penetration | High (lipophilic) | Low (P-glycoprotein substrate) |
| Sedation | Marked | Minimal |
| Anticholinergic effects | Significant | Minimal |
| Duration of action | 4-6 hours | 12-24 hours |
| Selectivity | Less H1-selective | More H1-selective |
| Examples | Diphenhydramine, chlorpheniramine, promethazine | Cetirizine, loratadine, fexofenadine |
3. First-Generation H1 Antihistamines (by Chemical Class)
A. Ethanolamines - Prototype: Diphenhydramine (Benadryl)
- Strong anticholinergic + sedative effects
- ~50% of patients get significant somnolence
- Low GI side effects
- Uses: Allergic reactions, motion sickness, insomnia, anaphylaxis (adjunct), Parkinsonism (anti-muscarinic benefit)
- Other members: Clemastine, dimenhydrinate (for motion sickness)
B. Ethylenediamines - Prototype: Pyrilamine (Mepyramine)
- Among the most H1-selective of the first-generation drugs
- Relatively weak CNS effects, but some sedation
- GI side effects are common
- Other members: Tripelennamine
C. Alkylamines - Prototype: Chlorpheniramine (Chlor-Trimeton)
- Among the most potent H1 antagonists
- Less sedation than ethanolamines - better for daytime use
- More CNS stimulation side effects than other groups
- Uses: Allergic rhinitis, urticaria, common cold formulations
- Other members: Brompheniramine, dexchlorpheniramine (active enantiomer)
D. Piperazines (First-Generation) - Prototype: Hydroxyzine (Atarax/Vistaril)
- Long-acting compound
- Prominent CNS depression - contributes to antipruritic action
- Uses: Skin allergies (urticaria, atopic dermatitis), anxiety, pre-operative sedation
- Cyclizine and meclizine - primarily for motion sickness
E. Phenothiazines - Prototype: Promethazine (Phenergan)
- Significant sedative + strong anticholinergic
- Also a dopamine (D2) receptor blocker
- Uses: Antiemetic (primary use), pre-operative sedation, motion sickness, adjunct to opioid analgesia
- Warning: Not recommended in children under 2 years (risk of fatal respiratory depression)
F. Piperidines (First-Generation) - Prototype: Cyproheptadine (Periactin)
- Uniquely has both H1 antihistamine AND 5-HT2A antiserotonin activity
- Causes drowsiness + significant anticholinergic effects
- Stimulates appetite (used clinically for appetite stimulation, anorexia)
- Uses: Urticaria, pruritus, serotonin syndrome (5-HT2A antagonism), appetite stimulation, migraine prophylaxis
4. Second-Generation H1 Antihistamines
These agents were developed starting in the 1980s to overcome the sedation and anticholinergic liabilities of the first generation.
A. Piperidines (Second-Generation) - Loratadine, Fexofenadine, Desloratadine
Loratadine (Claritin)
- Prodrug - metabolized to active desloratadine
- Non-sedating, no anticholinergic effects
- Poor CNS penetration (P-gp substrate)
- Uses: Allergic rhinitis, chronic urticaria
Fexofenadine (Allegra)
- Active metabolite of terfenadine (which was withdrawn for torsade de pointes risk)
- Lacks the cardiac toxicity of terfenadine
- Does not penetrate CNS
- Uses: Seasonal allergic rhinitis, urticaria
Desloratadine (Clarinex)
- Active metabolite of loratadine; slightly more potent
- Mast cell-stabilizing + anti-inflammatory properties
- Uses: Allergic rhinitis, urticaria
Historical note: Terfenadine and astemizole (early 2nd-gen) were withdrawn from the market because they blocked cardiac hERG K⁺ channels → prolonged QT → torsade de pointes. This is especially dangerous when combined with CYP3A4 inhibitors (azole antifungals, macrolide antibiotics) that increase their blood levels.
B. Piperazines (Second-Generation) - Cetirizine, Levocetirizine
Cetirizine (Zyrtec)
- Metabolite of hydroxyzine
- Minimal anticholinergic effects, low CNS penetration
- Slightly more sedating than other 2nd-gen agents (still much less than 1st gen)
- Additional mast cell-stabilizing and anti-inflammatory properties
- Uses: Allergic rhinitis, chronic urticaria, atopic dermatitis
Levocetirizine (Xyzal)
- Active R-enantiomer of cetirizine
- ~2x more potent; used at half the dose
- Less sedation than racemic cetirizine
- Mast cell-stabilizing properties
C. Alkylamine (Second-Generation) - Acrivastine
- Derivative of triprolidine (first-gen)
- Slightly more sedating than other 2nd-gen agents
D. Dibenzoxepine Tricyclic (Topical) - Olopatadine (Patanol, Pataday)
- Topical H1 antagonist + mast cell stabilizer + anti-inflammatory
- Available as eye drops (allergic conjunctivitis) and nasal spray (allergic rhinitis)
- Minimal systemic absorption - essentially no systemic side effects
5. H2 Antihistamines (Antisecretory)
These block H2 receptors on gastric parietal cells - completely different clinical role from H1 blockers.
| Drug | Notes |
|---|---|
| Cimetidine | First H2 blocker; significant CYP450 inhibitor - many drug interactions; weak anti-androgenic effects |
| Ranitidine | Withdrawn in 2020 (NDMA contamination concern) |
| Famotidine | Most potent H2 blocker; minimal drug interactions; preferred agent |
| Nizatidine | Similar to ranitidine; also withdrawn in many markets |
Mechanism: Competitive antagonism at H2 receptors → ↓cAMP in parietal cells → ↓H⁺/K⁺-ATPase activity → ↓gastric acid secretion
Clinical uses: Peptic ulcer disease, GERD, Zollinger-Ellison (as adjunct), stress ulcer prophylaxis, dyspepsia
Note: H2 blockers have been largely superseded by PPIs (proton pump inhibitors) for acid-related disease, but still used for mild GERD and on-demand relief.
6. H3 and H4 Receptor Antagonists (Emerging)
H3 antagonists:
- Pitolisant (Wakix) - FDA approved for narcolepsy (increases histamine release in CNS → promotes wakefulness)
- Investigational in ADHD, Alzheimer's, obesity
H4 antagonists:
- None FDA-approved yet
- Promising for allergic rhinitis, asthma, atopic dermatitis, pruritus, rheumatoid arthritis
7. Pharmacokinetics Summary
| Parameter | 1st Generation | 2nd Generation |
|---|---|---|
| Onset | 15-30 min (oral) | 1-3 hours |
| Duration | 4-6 hours | 12-24 hours |
| Protein binding | Moderate | High (fexofenadine lower) |
| Metabolism | Hepatic (CYP) | Hepatic / renal (fexofenadine - mostly renal excretion unchanged) |
| CNS penetration | Yes | No (P-gp efflux) |
8. Clinical Uses (Consolidated)
| Indication | Drug of Choice | Notes |
|---|---|---|
| Seasonal allergic rhinitis | Cetirizine, loratadine, fexofenadine | 2nd gen preferred (non-sedating) |
| Chronic urticaria | Cetirizine, fexofenadine | Higher doses sometimes needed |
| Acute urticaria / anaphylaxis | Diphenhydramine (IV/IM) | Adjunct; epinephrine is primary |
| Atopic dermatitis (pruritus) | Hydroxyzine (sedating), cetirizine | Nocturnal sedation is sometimes desired |
| Motion sickness | Promethazine, dimenhydrinate, meclizine | Promethazine most effective |
| Nausea/vomiting | Promethazine | Also blocks D2 |
| Pre-operative sedation | Promethazine, hydroxyzine | |
| Insomnia (OTC) | Diphenhydramine, doxylamine | Short-term only; tolerance develops rapidly |
| Peptic ulcer / GERD | Famotidine (H2 blocker) | Usually combined with or replaced by PPI |
| Appetite stimulation | Cyproheptadine | 5-HT2A + H1 antagonism |
| Serotonin syndrome | Cyproheptadine | 5-HT2A antagonism |
| Allergic conjunctivitis | Olopatadine drops | Topical; excellent local effect |
| Parkinsonism | Diphenhydramine, orphenadrine | Anticholinergic mechanism |
| Narcolepsy | Pitolisant (H3 antagonist) | Promotes wakefulness |
9. Adverse Effects
First-Generation
- CNS: Sedation, impaired cognition, paradoxical excitation (especially in children)
- Anticholinergic: Dry mouth, urinary retention, blurred vision, constipation, tachycardia
- GI: Nausea, epigastric distress (ethylenediamines)
- CV: QT prolongation risk (terfenadine, astemizole - now withdrawn)
Second-Generation
- Minimal sedation (cetirizine slightly more than others)
- Minimal anticholinergic
- No significant QT prolongation risk at therapeutic doses
- Headache, fatigue, dry mouth (mild)
H2 Blockers
- Cimetidine: Gynecomastia, impotence, confusion (elderly), many drug interactions (CYP1A2, 2C9, 2D6, 3A4 inhibition)
- Generally well tolerated as a class
10. Important Drug Interactions
- Cimetidine inhibits multiple CYP enzymes - interacts with warfarin, phenytoin, theophylline, tricyclic antidepressants
- 1st-gen antihistamines + CNS depressants (alcohol, benzodiazepines, opioids) - additive sedation
- CYP3A4 inhibitors (ketoconazole, erythromycin) historically raised terfenadine/astemizole levels → torsade de pointes (reason for withdrawal)
- Anticholinergic additive effects with other antimuscarinic drugs
11. Special Populations
Pregnancy:
- Chlorpheniramine and diphenhydramine - traditionally considered relatively safe (category B)
- Loratadine and cetirizine - preferred 2nd-gen options in pregnancy
Elderly:
- Avoid 1st-gen antihistamines (Beers Criteria) - risk of confusion, falls, urinary retention, constipation
- Use 2nd-gen at lowest effective dose
Children:
- Promethazine contraindicated <2 years (fatal respiratory depression risk)
- Cetirizine and loratadine safe and approved for children ≥2 years
Renal impairment:
- Fexofenadine, cetirizine, levocetirizine - dose reduction needed (renal excretion)
Quick Memory Mnemonic
"CAP FLD" for H1 antagonist classes:
- C = Chlorpheniramine (alkylamine) - most potent, least sedating 1st-gen
- A = Anti-muscarinic (diphenhydramine) - most sedating
- P = Promethazine (phenothiazine) - antiemetic
- F = Fexofenadine - no sedation, no interaction
- L = Loratadine - once daily, non-sedating
- D = Desloratadine/cetirizine - 2nd gen standard
- Goodman & Gilman's The Pharmacological Basis of Therapeutics (Chapter 43), pp. 880-884
- Stahl's Essential Psychopharmacology (Histamine chapter)
Explain me pancreatitis from basics to clinical along with management and treatment
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a complete, textbook-graded guide to pancreatitis from basics to clinical management:
Pancreatitis: Basics to Clinical Management
Based on Schwartz's Principles of Surgery (11th ed.), Robbins & Kumar Basic Pathology, and Sleisenger & Fordtran's GI and Liver Disease.
Part 1: Anatomy Basics (The Foundation)
The pancreas is a retroperitoneal organ, 75-100 g, 15-20 cm long, sloping from the C-loop of the duodenum to the splenic hilum. It has four parts: head, neck, body, and tail.
- The neck lies over L1-L2 - blunt trauma compresses it against the spine, causing ductal injury
- The tail sits in the splenic hilum - vulnerable during splenectomy or left colectomy
- The duct of Wirsung (main duct) + common bile duct join at the ampulla of Vater, draining into the duodenum
- Pancreas divisum (failed ventral-dorsal fusion) is the most common congenital anomaly and a risk factor for pancreatitis
Why Doesn't the Pancreas Digest Itself Normally?
Four key protective mechanisms:
- Enzymes made as inactive zymogens (trypsinogen, chymotrypsinogen, proelastase)
- Zymogens stored in separate granules from lysosomal hydrolases
- SPINK1 - a trypsin inhibitor inside acinar cells
- Trypsin self-inactivates by cleaving itself (negative feedback loop)
Pancreatitis occurs when these protections are overwhelmed.
Part 2: Acute Pancreatitis
Definition
A reversible inflammatory disorder ranging from focal edema to widespread hemorrhagic necrosis. If the cause is removed, function returns to normal.
- Incidence: 33-74 per 100,000 globally
- Overall mortality: ~5%; rising to 30-50% in severe disease
- 80% mild and self-limiting; 20% develop severe disease
Etiology - "I GET SMASHED"
| Letter | Cause | Key Details |
|---|---|---|
| I | Idiopathic | 10-20%; many have occult genetic basis |
| G | Gallstones | Most common in US; impaction at ampulla of Vater |
| E | Ethanol | Second most common; multiple mechanisms |
| T | Trauma | Blunt abdominal injury to neck of pancreas |
| S | Steroids | Especially in children; thiazides also |
| M | Mumps/Infections | Coxsackievirus, CMV, Ascaris, Clonorchis |
| A | Autoimmune | IgG4-related autoimmune pancreatitis |
| S | Scorpion sting | Tityus trinitatis |
| H | Hypertriglyceridemia/Hypercalcemia | TG >1000 mg/dL = 5-10% of cases; hyperparathyroidism |
| E | ERCP | Post-ERCP pancreatitis in 5-10% |
| D | Drugs | Azathioprine, furosemide, estrogens, propofol |
Gallstones + Alcohol together account for ~80% of all acute pancreatitis cases.
Pathophysiology
The central event is premature, intrapancreatic activation of digestive zymogens - autodigestion, as proposed by Chiari in 1896.
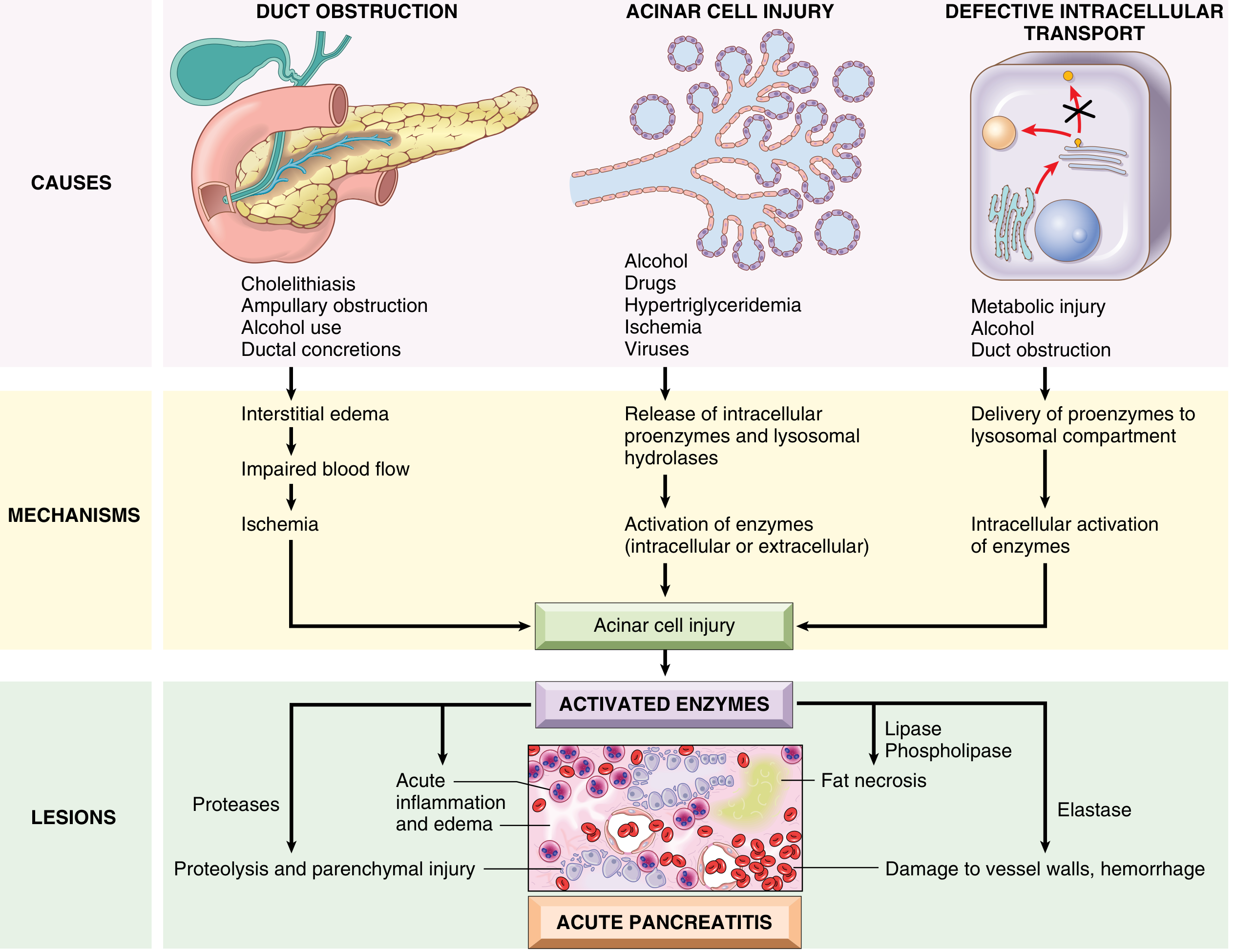
Fig: Pathogenesis of Acute Pancreatitis (Robbins & Kumar)
Three Initiating Pathways:
1. Duct Obstruction (gallstone, biliary sludge, tumor)
- ↑intraductal pressure → enzyme-rich interstitial accumulation
- Lipase (secreted already active) → immediate fat necrosis
- Injured tissue releases cytokines → edema → ischemia → more injury
2. Primary Acinar Cell Injury (alcohol, hypertriglyceridemia, ischemia, viruses)
- Alcohol: ↑exocrine secretion + sphincter of Oddi contraction → outflow obstruction + direct toxicity; promotes protein plug formation in ducts
- Hypertriglyceridemia: chylomicrons retard capillary flow → ischemia → lipase release → toxic free fatty acids
- Direct viral acinar cell infection
3. Defective Intracellular Transport (metabolic injury, alcohol, duct obstruction)
- Digestive proenzymes co-packaged with lysosomal hydrolases (cathepsin B)
- Intracellular trypsinogen activation → lysosomal rupture → cascading enzyme release
The Trypsin Cascade - "Master Activator":
Once trypsin is activated, it activates everything else:
- Other zymogens: chymotrypsin, elastase, phospholipase A2
- Kinin system (via kallikrein) → vasodilation + ↑permeability
- Clotting cascade (via factor XII/Hageman) → DIC
- Complement system → systemic SIRS
- Elastase → vessel wall destruction → hemorrhage
- Phospholipase A2 → damages surfactant → ARDS
- Lipase → fat necrosis → calcium soap precipitation → hypocalcemia
Morphology
Mild - Interstitial Edematous Pancreatitis:
- Interstitial edema
- Focal fat necrosis (yellow-white chalky deposits = calcium soaps)
- Preserved architecture, no vascular injury
Severe - Acute Necrotizing Pancreatitis:
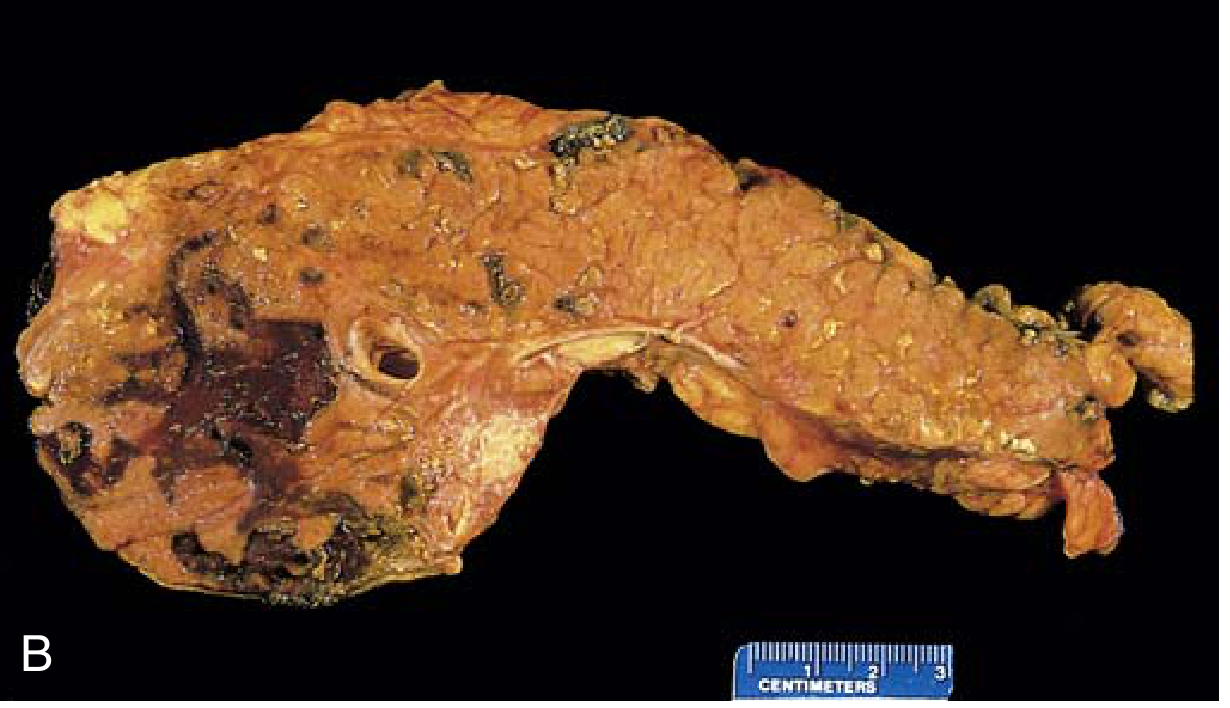
Fig: Acute necrotizing pancreatitis - dark hemorrhagic zones + yellow-white fat necrosis (Robbins)
- Damage extends to acini, ducts, islets, and blood vessels
- Peritoneal fluid: serous, brown-tinged, with fat globules
- Fat necrosis can extend to omentum, mesentery, even subcutaneous fat
- Histology: neutrophilic infiltration, acinar cell necrosis, vascular injury, hemorrhage
Clinical Presentation
Symptoms:
- Epigastric pain - sudden, severe, constant, bores through to the back (mid-thoracic)
- Nausea and vomiting (does not relieve pain - distinguishes from bowel obstruction)
- Low-grade fever
Signs:
- Epigastric tenderness ± guarding, rigidity
- Absent or reduced bowel sounds (paralytic ileus)
- Cullen's sign - periumbilical bruising (retroperitoneal hemorrhage tracking forward)
- Grey Turner's sign - flank bruising (retroperitoneal hemorrhage tracking laterally)
- Jaundice if gallstone obstructs CBD
- Shock in severe cases (hypovolemia + vasodilation)
Investigations
Serum Enzymes:
| Test | Rise | Peak | Returns to Normal | Notes |
|---|---|---|---|---|
| Amylase | 6-12 hrs | 24 hrs | 3-5 days | Sensitivity ~80%, non-specific; can be normal in severe necrosis |
| Lipase | 4-8 hrs | 24 hrs | 7-14 days | More sensitive + specific; preferred marker |
Diagnosis: lipase or amylase >3x upper limit of normal
Other Labs:
- Leukocytosis, ↑glucose, ↑BUN/Cr (hypovolemia)
- ↑ALT >3x = strongly suggests gallstone etiology (>95% PPV in right clinical context)
- ↓Ca²⁺ = saponification = severe disease / poor prognosis
- ↑TG if hypertriglyceridemia is cause
- CRP >150 mg/L at 48 hours = marker of severity
Imaging:
| Modality | Role |
|---|---|
| Ultrasound | First-line; gallstones, biliary dilation; poor pancreatic visualization due to bowel gas |
| CT with contrast (CECT) | Gold standard for severity; detects necrosis (enhancement failure), fluid collections, abscess |
| MRI/MRCP | Duct anatomy, choledocholithiasis, no radiation |
| ERCP | Therapeutic only; for persistent CBD stone + cholangitis |
| EUS | Detects microlithiasis, occult stones |
Severity Scoring
Revised Atlanta Classification (2012) - Current Standard:
| Grade | Criteria |
|---|---|
| Mild | No organ failure, no local complications; resolves within 1 week |
| Moderately Severe | Transient organ failure (<48 hrs) AND/OR local complications |
| Severe | Persistent organ failure (>48 hrs), single or multiorgan |
Ranson's Criteria (11 factors):
At admission (GAL BUG):
- G - Glucose >200 mg/dL
- A - Age >55
- L - LDH >350 IU/L
- B - (W)BC >16,000
- U - (as)T >250 IU/L (= AST)
At 48 hours (HOBBS):
- H - Hematocrit drop >10%
- O - (BUN rise >5 = "Oh no, kidneys")
- B - Base deficit >4
- B - (Ca²⁺) <8 mg/dL
- S - Sequestration of fluid >6 L; PaO₂ <60 mmHg
| Score | Mortality |
|---|---|
| 0-2 | <1% |
| 3-4 | ~16% |
| 5-6 | ~40% |
| >6 | ~100% |
BISAP Score (≥3 = severe):
- BUN >25, Impaired mental status, SIRS, Age >60, Pleural effusion
CT Severity Index (Balthazar + Necrosis score):
- Score >6 = severe (mortality 17%, morbidity 92%)
Local Complications (Atlanta 2012 Definitions):
| Complication | Timing | Wall | Contents | Management |
|---|---|---|---|---|
| Acute peripancreatic fluid collection | <4 weeks | No wall | Fluid only | Mostly resolve spontaneously |
| Pancreatic pseudocyst | >4 weeks | Defined wall (no epithelium) | Fluid | Observe if asymptomatic; drain if symptomatic |
| Acute necrotic collection | <4 weeks | No wall | Necrosis ± fluid | Antibiotics; defer intervention |
| Walled-off necrosis (WON) | >4 weeks | Defined wall | Necrosis ± fluid | Step-up approach if infected |
Pseudocyst - lined by fibrin and granulation tissue (NOT epithelium) - develops in ~10% of acute pancreatitis cases and in chronic pancreatitis.
Systemic Complications:
- ARDS - phospholipase A2 damages alveolar surfactant
- AKI - hypovolemia + inflammation
- DIC - trypsin activates factor XII + clotting cascade
- Shock - third spacing of fluid, systemic vasodilation
- Hyperglycemia - islet cell destruction
- Hypocalcemia - fat saponification
- Ileus, pancreatic ascites, left-sided pleural effusion
Part 3: Treatment and Management of Acute Pancreatitis
Step 1: Fluid Resuscitation (MOST IMPORTANT early intervention)
- Lactated Ringer's solution preferred over normal saline (reduces SIRS in studies)
- Goal: restore normal BP, heart rate, urine output >0.5 mL/kg/hr
- Moderate resuscitation (avoid excessive fluid in elderly/cardiac/renal patients)
- Monitor with BUN trends (rising BUN = inadequate resuscitation)
Step 2: Analgesia
- IV opioids (fentanyl, hydromorphone, morphine)
- Historical avoidance of morphine (sphincter of Oddi concern) is no longer clinically relevant
- Epidural analgesia in severe cases
Step 3: Nutrition
- Mild pancreatitis: Early oral feeding as soon as tolerated (low-fat soft diet)
- Severe pancreatitis: Early enteral nutrition within 24-48 hours (nasogastric or nasojejunal tube)
- Enteral > TPN: maintains gut barrier, reduces bacterial translocation, fewer infectious complications, lower cost
- TPN only if enteral route is impossible
Step 4: Antibiotics
- NOT routine - multiple RCTs showed no benefit for prophylactic antibiotics
- Use ONLY for:
- Confirmed infected pancreatic necrosis (gas on CT or positive cultures)
- Associated cholangitis
- Other proven infections
- Agent: imipenem/meropenem (best pancreatic penetration)
Step 5: Treat the Cause
| Etiology | Treatment |
|---|---|
| Gallstones + cholangitis/CBD obstruction | Urgent ERCP within 24-72 hours |
| Gallstones (mild, resolving) | Cholecystectomy same admission or within 2-4 weeks |
| Alcohol | Counseling + cessation |
| Hypertriglyceridemia | Insulin drip ± plasmapheresis if TG >1000 |
| Drugs | Stop offending drug |
| Hypercalcemia | Treat hyperparathyroidism |
Step 6: Monitoring (ICU for Severe Cases)
- Continuous monitoring of vitals, urine output, SpO₂
- Serial labs: BUN, Cr, Ca²⁺, CBC, CRP, LFTs
- Daily organ failure assessment
- CECT at 48-72 hours if clinical deterioration
Management of Infected Necrosis - Step-Up Approach (Current Standard)
- Antibiotics (carbapenem) + NPO + supportive care
- If no improvement at 72-96 hours: percutaneous CT-guided drainage or endoscopic transluminal drainage (transgastric)
- If still failing: minimally invasive necrosectomy (video-assisted retroperitoneal debridement - VARD, or endoscopic necrosectomy)
- Open surgical necrosectomy - last resort; highest morbidity
Key principle: Wait until necrosis is walled off (>4 weeks) before intervention if clinically possible - margins are better defined, bleeding risk is lower.
Part 4: Chronic Pancreatitis
Definition
An irreversible, progressive inflammatory condition causing permanent destruction of exocrine parenchyma, eventually affecting endocrine tissue. Unlike acute pancreatitis, damage does not reverse.
Etiology - TIGAR-O
- Toxic-metabolic: alcohol, tobacco, hypercalcemia, hypertriglyceridemia, chronic renal failure
- Idiopathic: 15-25%
- Genetic: PRSS1, SPINK1, CFTR mutations
- Autoimmune: IgG4-related
- Recurrent severe acute pancreatitis
- Obstructive: pancreas divisum, strictures, tumors
Genetics of Hereditary Pancreatitis
- PRSS1 gene (cationic trypsinogen, chromosome 7q35): R122H and N291 mutations = gain-of-function → trypsin cannot self-inactivate → persistent autodigestion
- SPINK1 mutations: loss-of-function trypsin inhibitor → uncontrolled trypsin activity
- CFTR mutations: defective bicarbonate/fluid secretion → viscous secretions → duct plugging
- Autosomal dominant, 80% penetrance; presents in childhood
- Lifetime pancreatic cancer risk: 40% in PRSS1 hereditary pancreatitis
Pathology
- Extensive fibrosis replacing acinar tissue
- Acinar atrophy → exocrine insufficiency
- Islets preserved initially, then destroyed → Type 3c diabetes
- Ductal calcifications (protein plugs calcify) - pathognomonic on imaging
- Dilated ducts ("chain of lakes" on MRCP)
Clinical Features
- Chronic epigastric pain radiating to back; worse with eating + alcohol; improves leaning forward
- Steatorrhea - foul-smelling, floating, fatty stools; requires >90% exocrine destruction to manifest
- Weight loss and malabsorption
- Fat-soluble vitamin deficiency (A, D, E, K) → osteoporosis (Vitamin D), coagulopathy (Vitamin K)
- Type 3c (pancreatogenic) diabetes - brittle, prone to hypoglycemia (lost glucagon from alpha cells too)
- Jaundice (CBD stricture from fibrosis)
- Episodes of acute-on-chronic exacerbations
Investigations
- Amylase/lipase may be normal in burned-out disease (insufficient acinar tissue)
- CT: pancreatic calcifications, ductal dilation, atrophy
- MRCP: ductal irregularity, strictures, "chain of lakes"
- Fecal elastase-1 <200 μg/g = exocrine insufficiency
- Secretin stimulation test = gold standard for exocrine function
- Glucose tolerance test for diabetes
Treatment of Chronic Pancreatitis
1. Lifestyle:
- Absolute alcohol abstinence
- Smoking cessation
- Low-fat diet (5-6 small meals/day)
2. Pain Control:
- WHO ladder: paracetamol/NSAIDs → tramadol → strong opioids
- Pancreatic enzyme supplements - reduce CCK-driven pancreatic stimulation → pain relief
- Pregabalin, tricyclic antidepressants - centrally-mediated pain
- Celiac plexus block (EUS-guided) - temporary relief
3. Pancreatic Enzyme Replacement Therapy (PERT):
- Pancrelipase (high lipase content) with every meal and snacks
- Fat-soluble vitamins supplementation (A, D, E, K)
- Goal: eliminate steatorrhea, restore normal nutrition
4. Diabetes (Type 3c) Management:
- Insulin (usually required in advanced disease)
- Avoid sulfonylureas - risk of severe hypoglycemia
- Monitor carefully (brittle hypoglycemia risk)
5. Endoscopic Therapy:
- Ductal stenting for dominant strictures
- ESWL (extracorporeal shock wave lithotripsy) for calculi → then endoscopic stone removal
- Pancreatic sphincterotomy
6. Surgery - Indications:
- Intractable pain unresponsive to medical/endoscopic therapy
- Duodenal or biliary obstruction
- Suspicion of malignancy
- Pancreatic fistula
Surgical Procedures:
| Type | Procedure | Indication |
|---|---|---|
| Drainage | Puestow procedure (lateral pancreaticojejunostomy) | Dilated duct >6 mm |
| Drainage + decompression | Frey procedure (ductal drainage + head coring) | Dilated duct + head-predominant |
| Resection | Whipple (pancreaticoduodenectomy) | Head disease, non-dilated duct, mass |
| Resection | Beger (duodenum-preserving head resection) | Head disease, preserves duodenum |
| Resection | Distal pancreatectomy | Body/tail disease |
| Radical | Total pancreatectomy + islet autotransplantation (TPIAT) | Refractory hereditary pancreatitis |
Summary Comparison
| Feature | Acute Pancreatitis | Chronic Pancreatitis |
|---|---|---|
| Reversibility | Reversible | Irreversible |
| Most common cause | Gallstones (West) | Alcohol (West) |
| Pain character | Sudden, severe, boring to back | Chronic, post-meal, back radiation |
| Enzyme levels | Markedly elevated | May be normal (burned-out) |
| Exocrine insufficiency | Not usually | Yes - steatorrhea |
| Endocrine insufficiency | Transient hyperglycemia | Type 3c diabetes |
| Calcifications on CT | Rare | Yes - pathognomonic |
| Mortality | ~5% overall; 30-50% severe | 50% over 20-25 years |
| Cancer risk | Low | Elevated (40% in PRSS1 hereditary) |
10 High-Yield Clinical Pearls
- Lipase > Amylase as the diagnostic test - more specific, stays elevated longer
- ALT >3x = gallstone etiology with >95% positive predictive value
- Cullen + Grey Turner signs = retroperitoneal hemorrhage = necrotizing pancreatitis
- Hypocalcemia = fat saponification = poor prognostic sign
- ERCP in gallstone pancreatitis only if cholangitis or persistent CBD obstruction - not routine
- Enteral > Parenteral nutrition in severe pancreatitis - start within 24-48 hours
- No prophylactic antibiotics in acute pancreatitis - multiple RCTs showed no benefit
- Step-up approach for infected necrosis; wait for walled-off necrosis (>4 weeks) before intervening
- Amylase normal in burned-out chronic pancreatitis - use fecal elastase instead
- PRSS1 hereditary pancreatitis = 40% lifetime risk of pancreatic cancer - long-term surveillance required
Sources:
- Schwartz's Principles of Surgery, 11th ed., Chapter 33, pp. 1439-1468
- Robbins & Kumar Basic Pathology, Chapter 15, pp. 640-646
- Sleisenger & Fordtran's Gastrointestinal and Liver Disease
This is a shared conversation. Sign in to Orris to start your own chat.