Metabolic myopthies
Reading File
Finding Sources
Searching PubMed
"metabolic myopathy" diagnosis management
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
metabolic myopathy ragged red fibers mitochondrial glycogen storage muscle biopsy
Now I have comprehensive material. Let me compile a thorough response.
Metabolic Myopathies
Metabolic myopathies are a group of hereditary (and occasionally acquired) muscle disorders in which defects in biochemical pathways that generate ATP lead to exercise intolerance, muscle pain, contractures, and myoglobinuria. They are classically divided into three major categories: disorders of carbohydrate metabolism, disorders of lipid metabolism, and disorders of mitochondrial function.
Core Pathophysiology
The fundamental defect in all metabolic myopathies is impairment of ATP synthesis in skeletal muscle. Normally, muscle has safety mechanisms that prevent exercise to the point of destruction. In metabolic myopathies, these mechanisms fail:
- Forced exercise → muscle pain → contracture (electrically silent, unlike a cramp)
- Continued exertion → rhabdomyolysis → myoglobinuria → risk of acute tubular necrosis
- Symptoms span: fatigue → muscle pain → contractures → myoglobinuria (escalating severity)
"In the metabolic muscle diseases, maintenance of ATP levels is impaired, and the protective mechanism that functions in the normal person is absent." — Bradley and Daroff's Neurology in Clinical Practice
1. Disorders of Carbohydrate Metabolism
Glycogen provides the dominant fuel in the early (first ~20 min) phase of exercise. Defects in glycolysis cause symptoms that arise rapidly with intense, short-duration effort.
McArdle Disease (Myophosphorylase Deficiency — Type V GSD)
- Gene/locus: PYGM, chromosome 11q13; autosomal recessive
- Defect: Absence of myophosphorylase → muscle cannot liberate glucose from glycogen
- Clinical: Exercise intolerance, muscle pain, and contractures within minutes of vigorous activity; classic "second-wind" phenomenon (symptoms improve after ~8–10 min as the body switches to circulating metabolites)
- Lab: CK elevated (markedly during/after attacks); no rise in venous lactate after ischemic forearm exercise (flat lactate test)
- Biopsy: Subsarcolemmal accumulation of glycogen; absent myophosphorylase on histochemistry
- Treatment: Aerobic training (promotes second wind); pre-exercise sucrose supplementation
Pompe Disease (Acid Maltase Deficiency — Type II GSD)
- Gene/locus: GAA; autosomal recessive
- Defect: Deficiency of lysosomal acid α-glucosidase → glycogen accumulates in lysosomes → lysosomal rupture → myofiber destruction
- Three clinical forms:
| Form | Onset | Features |
|---|---|---|
| Infantile (classic) | < 6 months | Hypotonia, cardiomegaly, macroglossia, hepatomegaly; fatal by age 2 (cardiorespiratory failure) |
| Juvenile | First decade | Progressive proximal weakness, respiratory involvement; resembles Duchenne or LGMD |
| Adult-onset | 3rd–4th decade | Proximal > distal weakness; predominant respiratory muscle involvement (diaphragm); resembles polymyositis or LGMD |
- Treatment: Enzyme replacement therapy (ERT) with alglucosidase alfa; newer cipaglucosidase alfa + miglustat (chaperone) has shown improved outcomes in late-onset Pompe disease (meta-analysis, PMID 41757410)
Other Glycolytic Enzyme Deficiencies
| Enzyme | Key Features |
|---|---|
| PFK deficiency (Type VII — Tarui disease) | Clinically identical to McArdle; no second-wind; mild hemolytic anemia (red cells also lack M-subunit of PFK); nausea/vomiting with attacks |
| Phosphoglycerate kinase deficiency | X-linked (Xq13); may include hemolytic anemia, intellectual disability, seizures |
| Phosphoglycerate mutase deficiency | Muscle pain + myoglobinuria; elevated uric acid |
| Lactate dehydrogenase deficiency | High CK but low LDH (discrepancy); pyruvate rises after exercise but lactate does not |
| β-Enolase deficiency | Exercise intolerance, myalgia, episodic hyperCKemia |
2. Disorders of Lipid Metabolism
Fatty acids become the dominant fuel after ~20–30 minutes of sustained aerobic exercise. Defects here cause symptoms during prolonged, endurance-type exertion — not brief sprints.
Carnitine Palmitoyltransferase II (CPT-II) Deficiency
- Most common disorder of lipid metabolism causing episodic muscle disease
- Mechanism: CPT-I (outer mitochondrial membrane) links carnitine to long-chain fatty acids; CPT-II (inner membrane) unlinks it. Deficiency → fatty acids cannot enter mitochondria
- Clinical: Myoglobinuria after prolonged exercise (mountain climbing, tennis); attacks worsened by fasting, infection, or cold; patients often muscular (prefer sprinting/weightlifting)
- Lab: CK normal between attacks; ischemic forearm exercise test is normal (glycolytic pathway intact); RER remains near 1.0 even at rest
- Biopsy: Normal between attacks
- Treatment: Avoid prolonged fasting + strenuous exercise; glucose supplementation during exercise; admit for forced alkaline diuresis if myoglobinuria occurs
Other Lipid Storage Disorders
- Primary carnitine deficiency: Cardiomyopathy + proximal myopathy; treated with oral carnitine
- Neutral lipid storage disease (NLSD):
- NLSD with myopathy (NLSD-M): Due to mutations in ATGL (adipose triglyceride lipase) — progressive myopathy
- NLSD with ichthyosis (NLSD-I / Chanarin-Dorfman syndrome): Mutations in CGI-58 (co-activator of ATGL); associated with skin lesions
3. Mitochondrial Myopathies
The most complex category, with dual (nuclear + mitochondrial) genetic control. Mitochondrial mutations show maternal inheritance and heteroplasmy (variable disease expression based on threshold of mutant mtDNA).
Pathophysiology
- Both fatty acids and glycolysis converge on acetyl-CoA → Krebs cycle → electron transport chain (ETC) → ATP
- mtDNA encodes 13 ETC proteins + 22 tRNAs + 2 rRNAs; rest are nuclear-encoded
- When mitochondrial function is impaired, resting muscle may require fully activated oxidative metabolism → symptoms even at rest; anaerobic backup → lactic acidosis
Morphology
- Ragged red fibers (RRF): Pathognomonic — subsarcolemmal accumulation of abnormal mitochondria; seen on modified Gomori trichrome stain (red blotches)
- COX-negative / SDH-positive fibers on sequential histochemistry
- Electron microscopy: Structurally abnormal mitochondria with paracrystalline inclusions
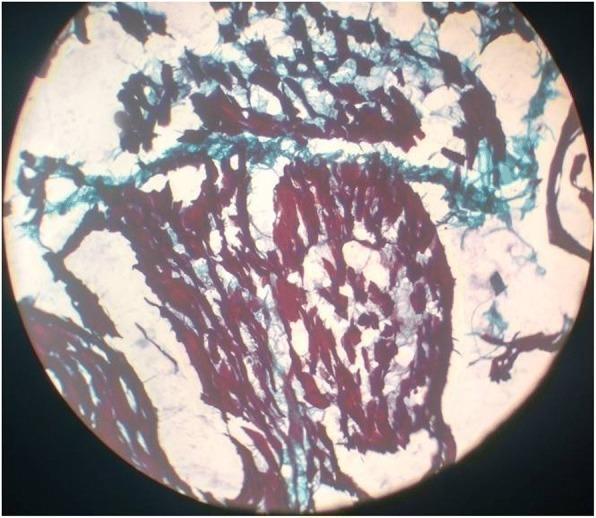
Major Syndromes
| Syndrome | Full Name | Key Features |
|---|---|---|
| MELAS | Mitochondrial Encephalopathy, Lactic Acidosis, Stroke-like Episodes | Most common (m.3243A>G in tRNA^Leu); proximal weakness + stroke-like episodes (especially posterior cortex), dementia, lactic acidosis, RRF |
| MERRF | Myoclonic Epilepsy with Ragged Red Fibers | Myoclonus + epilepsy + ataxia + RRF; m.8344A>G |
| CPEO/KSS | Chronic Progressive External Ophthalmoplegia / Kearns-Sayre Syndrome | Bilateral ptosis + ophthalmoplegia; KSS also has cardiac conduction defects, retinitis pigmentosa, cerebellar ataxia (onset < 20 yrs) |
| MILS | Maternally Inherited Leigh Syndrome | Subacute necrotizing encephalomyelopathy; regression, brainstem signs |
| NARP | Neuropathy, Ataxia, Retinitis Pigmentosa | m.8993T>G; ataxia + retinitis pigmentosa |
| MLASA | Mitochondrial Myopathy, Lactic Acidosis, Sideroblastic Anemia | Rare; caused by mutations in PUS1 or YARS2 |
Clinical Features of Mitochondrial Myopathies (General)
- Onset often in childhood; exercise → heavy limbs, muscle aches, nausea, breathlessness
- Fixed proximal weakness (may be sole presentation)
- Elevated resting lactate/pyruvate (L:P ratio >20 suggests ETC defect)
- Multi-system involvement: CNS, heart (cardiomyopathy, arrhythmia), eyes, ears (sensorineural deafness), endocrine (DM)
- Short stature, ptosis, ophthalmoplegia are common clues
4. Disorder of Nucleotide Metabolism
Myoadenylate Deaminase (AMPDA) Deficiency
- Affects ~1–2% of the population (most are asymptomatic)
- Enzyme converts AMP → IMP + ammonia; supports ATP recycling during exercise
- Forearm exercise test: Normal lactate rise but no ammonia production (diagnostic)
- Clinical significance debated — poor correlation between enzyme defect and symptoms
Diagnostic Approach
Investigations
| Test | Utility |
|---|---|
| Serum CK | Elevated during/after attacks; can be normal between attacks in CPT deficiency |
| Ischemic forearm exercise test | Glycolytic disorders: no lactate rise; AMPDA deficiency: no ammonia rise |
| Serum lactate/pyruvate | Elevated at rest or with minimal exercise in mitochondrial disorders |
| Urine myoglobin | Present during rhabdomyolytic attacks |
| EMG | Myopathic pattern; contractures are electrically silent |
| Muscle biopsy | Gold standard; histochemistry (PAS for glycogen, oil red O for lipid, Gomori trichrome for RRF, COX/SDH staining) |
| Enzyme assay (leukocytes/muscle) | Confirmatory for specific enzymatic defects |
| Genetic panel | Next-generation sequencing; mtDNA analysis for mitochondrial disorders |
Pattern Recognition
| Feature | Glycolytic Disorder | Lipid Disorder | Mitochondrial |
|---|---|---|---|
| Symptom trigger | Brief intense exercise | Prolonged aerobic exercise / fasting | Variable; rest → exertion |
| Second wind | Yes (McArdle) | No | No |
| Lactate with exercise | No rise (flat) | Normal | High at rest/low exercise |
| Biopsy finding | Glycogen accumulation | Lipid vacuoles | Ragged red fibers |
| Myoglobinuria | Common (McArdle, PFK) | Severe (CPT-II) | Occasional |
| Multi-system | Rare | Rare | Very common |
Management Summary
- Glycolytic disorders: Aerobic exercise training; sucrose supplementation pre-exercise (McArdle); avoid ischemia
- Lipid disorders: Avoid prolonged fasting; glucose supplementation; medium-chain triglyceride (MCT) diet in some; aggressive hydration if myoglobinuria
- Pompe disease: ERT (alglucosidase alfa); newer cipaglucosidase alfa + miglustat shows improved efficacy
- Mitochondrial: Largely supportive; CoQ10 supplementation (in CoQ10 deficiencies); manage complications (pacemaker for KSS, insulin for mitochondrial DM); avoid mitochondrial toxins (valproate, metformin in severe disease)
- All: Monitor renal function during myoglobinuric attacks; genetic counseling
Sources:
- Bradley and Daroff's Neurology in Clinical Practice, Chapters on Metabolic Myopathies
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Diseases of Lipid or Glycogen Metabolism + Mitochondrial Myopathies
- Robbins & Kumar Basic Pathology, Channelopathies, Metabolic Myopathies, and Mitochondrial Myopathies
- Recent meta-analysis on cipaglucosidase alfa + miglustat vs. alglucosidase alfa in late-onset Pompe disease [PMID: 41757410]
Explain forearm test and ammonia test
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Now I have everything needed for a thorough explanation.
Forearm Exercise Test (Non-Ischemic) & Ammonia Test
Background — Why This Test?
During brief, intense exercise, the forearm muscles rely almost entirely on anaerobic glycolysis (muscle glycogen → glucose → lactate). Two metabolic byproducts are generated in parallel:
- Lactate — end product of glycolysis via lactate dehydrogenase
- Ammonia (+ hypoxanthine, inosine) — produced via the purine nucleotide cycle:
- When ATP is consumed rapidly → ADP → AMP
- Adenylate kinase: 2 ADP → ATP + AMP
- AMP deaminase (AMPDA): AMP → IMP + NH₃ (ammonia)
Both pathways are activated simultaneously in normal muscle under anaerobic stress. This parallel relationship is the basis of the test.
The Test — Protocol
Classic (Ischemic) Forearm Exercise Test
- A blood pressure cuff is inflated above systolic pressure on the upper arm (creating ischemia)
- The patient grips a dynamometer repetitively for 1 minute at maximum effort
- The cuff is released; venous blood is drawn from the antecubital vein at baseline, then at 1, 3, 5, and 10 minutes post-exercise
Problem: Ischemic testing carries a risk of rhabdomyolysis in patients with glycolytic enzyme defects — the very patients being tested. It is now largely abandoned.
Non-Ischemic (Non-Occlusive) Forearm Exercise Test — Current Standard
- Same protocol but without the blood pressure cuff
- Exercise must be sufficiently strenuous that the forearm muscle outpaces blood-borne substrate delivery (effectively self-ischemic through high workload)
- Easy to perform, cost-effective, high sensitivity
- Venous samples drawn at the same intervals for lactate and ammonia
Normal Response
| Metabolite | Expected Rise |
|---|---|
| Lactate | ≥ 3× baseline (typically rises 3–5 fold) |
| Ammonia | ≥ 3× baseline |
Both rise together — this confirms:
- Glycolytic pathway is intact (lactate rises)
- Purine nucleotide cycle is intact (ammonia rises)
Interpretation of Abnormal Results
1. Glycolytic / Glycogenolytic Defect (e.g., McArdle disease, PFK deficiency, other glycolytic enzyme defects)
| Lactate | Ammonia |
|---|---|
| Flat — no rise | Normal or exaggerated rise |
Reason: Muscle cannot convert glycogen/glucose → pyruvate → lactate (glycolysis blocked). However, the purine nucleotide cycle is intact, so AMP is still deaminated to NH₃. In fact, because ATP depletion is excessive (glycolysis cannot regenerate it), ammonia may rise even more than normal.
"Patients with certain defects in the glycolytic pathways produce normal to excessive amounts of ammonia and hypoxanthine, but no lactate." — Bradley and Daroff's Neurology
Key point: In McArdle disease, exercise causes a painful electrically silent contracture — this distinguishes it from a cramp (which shows electrical activity on EMG).
2. Myoadenylate Deaminase (AMPDA) Deficiency
| Lactate | Ammonia |
|---|---|
| Normal rise | Flat — no rise |
Reason: The purine nucleotide cycle is broken at the AMP → IMP step, so no ammonia is produced. Glycolysis is entirely intact, so lactate rises normally.
This is the "ammonia test" — specifically diagnosing AMPDA deficiency by showing absent ammonia response with preserved lactate production.
3. Poor Effort / Non-Cooperative Patient
| Lactate | Ammonia |
|---|---|
| No rise | No rise |
Both are flat because the patient simply did not work hard enough to exceed the blood supply. This must be distinguished from a true metabolic defect — repeating the test with supervised effort is necessary.
4. Mitochondrial Myopathy
| Lactate | Ammonia/Hypoxanthine |
|---|---|
| Excessive rise at rest / low workload | Excessive rise |
Both are elevated disproportionately because mitochondrial failure forces excessive reliance on anaerobic metabolism even at low workloads — lactate piles up because the ETC cannot re-oxidize NADH efficiently.
Summary Table
| Condition | Lactate | Ammonia | Interpretation |
|---|---|---|---|
| Normal | ↑↑ | ↑↑ | Both pathways intact |
| McArdle / PFK / glycolytic defect | Flat | ↑↑ (normal or high) | Glycolysis blocked; purine cycle intact |
| AMPDA deficiency | ↑↑ (normal) | Flat | Purine cycle blocked; glycolysis intact |
| Poor effort | Flat | Flat | Neither pathway activated (no real exercise) |
| Mitochondrial myopathy | ↑↑↑ (excessive) | ↑↑↑ (excessive) | Anaerobic overload due to ETC failure |
Clinical Pearls
- The test targets forearm muscle specifically because it is anatomically convenient for venous sampling and creates a largely closed, self-contained muscular system during gripping exercise
- In CPT-II deficiency (lipid metabolism defect), the forearm test is normal — fatty acids aren't used in brief, intense exercise; symptoms only arise after prolonged aerobic effort
- The test is a screening tool — abnormal results require confirmation with muscle biopsy, enzyme assay, or genetic testing
- Forearm test is now largely replaced by genetic panel testing in most centers where molecular diagnostics are available, but it remains useful when targeted, inexpensive, and rapid screening is needed
Sources:
- Bradley and Daroff's Neurology in Clinical Practice, Exercise Testing
- Harrison's Principles of Internal Medicine 22E, Glycogen Storage and Glycolytic Defects
- Harrison's Principles of Internal Medicine 22E, Muscle Glycogen Storage Diseases
Approach to say in viva clinically for recurrent weakness chanalopathy vs myopathy vs nmj and also discuss hypokalemic vs hyperkalemic vs thyrotoxicosis vs Anderson Tamil vs mac ardle vs cpt2 vs mitochondrial clinically
Finding Sources
Finding Sources
Finding Sources
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.