Some Important topics: Leukaemia, anemia, purpura, hodgkin lymphoma, thalassemia, sickle cell anemia, DIC, blood grouping. Everyone please study these topics now
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
I now have rich content from the textbooks. Here is your complete study guide for all 8 important haematology topics:
📚 Haematology Study Guide - Important Topics
1. LEUKAEMIA
Definition: A malignant proliferation of white blood cell precursors originating in the bone marrow, classified by cell lineage and course.
Classification:
| Type | Cell of Origin | Course |
|---|---|---|
| ALL (Acute Lymphoblastic) | Lymphoid precursors | Acute |
| AML (Acute Myeloid) | Myeloid precursors | Acute |
| CLL (Chronic Lymphocytic) | Mature B cells | Chronic |
| CML (Chronic Myeloid) | Myeloid cells, BCR-ABL | Chronic |
Key Clinical Features:
- Symptoms: Fatigue, pallor, fever, bleeding, recurrent infections, bone pain
- Signs: Lymphadenopathy, splenomegaly, hepatomegaly
- Diagnosis: Blood film + bone marrow examination
Pulmonary Involvement (Leukaemia):
- Leukaemic infiltration found at autopsy in ~2/3 of patients
- Usually asymptomatic - rarely visible on CXR
- Leukostasis (in AML): WBC >100,000-300,000 cells/mm³ - leukaemic cells obliterate small pulmonary vessels causing dyspnoea
- T-cell leukaemias: massive mediastinal adenopathy that responds rapidly to chemo/radiotherapy
- Granulocytic sarcoma (chloroma): pleural mass of myeloid leukaemia cells - named for its green appearance
Robbins & Kumar Basic Pathology; Grainger & Allison's Diagnostic Radiology
2. ANAEMIA
Definition: Reduction in the oxygen-carrying capacity of blood, defined by haemoglobin below the normal range for age and sex.
Classification by Mechanism:
| Category | Causes |
|---|---|
| Decreased production | Iron deficiency, B12/folate deficiency, aplastic anaemia, bone marrow infiltration |
| Increased destruction (haemolytic) | Sickle cell, thalassaemia, autoimmune, G6PD deficiency |
| Blood loss | Acute or chronic haemorrhage |
Classification by MCV:
- Microcytic (MCV <80 fL): Iron deficiency, thalassaemia, chronic disease
- Normocytic (MCV 80-100 fL): Acute blood loss, haemolysis, chronic disease
- Macrocytic (MCV >100 fL): B12/folate deficiency, liver disease, hypothyroidism
Clinical Features:
- Pallor, fatigue, exertional dyspnoea, palpitations, tachycardia
- Severe: angina, heart failure
Pye's Surgical Handicraft; Bailey and Love's Short Practice of Surgery
3. PURPURA
Definition: Extravasation of red blood cells into the dermis - lesions do not blanch with pressure (this distinguishes from erythema).
Classification:
Non-palpable purpura (flat - due to bleeding disorders or vascular fragility):
- Trauma, solar/senile purpura, steroid purpura, stasis purpura
- Systemic causes: Thrombocytopenia (ITP), abnormal platelet function, clotting factor defects
Palpable purpura (raised - suggests vasculitis):
- Leukocytoclastic vasculitis, HSP (Henoch-Schonlein Purpura)
Important Systemic Causes:
| Mechanism | Examples |
|---|---|
| Clotting disturbance | ITP, haemophilia |
| Vascular fragility | Amyloidosis, Ehlers-Danlos, scurvy |
| Thrombi | DIC, purpura fulminans, warfarin necrosis, antiphospholipid syndrome |
| Emboli | Cholesterol, fat emboli |
| Infection | Meningococcaemia, SARS-CoV-2 |
ITP (Immune Thrombocytopenic Purpura):
- Antiplatelet IgG autoantibodies against platelet membrane glycoproteins
- Acute (children): follows viral infection, self-resolving
- Chronic (adults): persists >6 months; mainly females aged 15-50 years; associated with SLE
- Treatment: corticosteroids, IVIG, splenectomy
Harrison's Principles of Internal Medicine 22E; Andrews' Diseases of the Skin; Bailey and Love
4. HODGKIN LYMPHOMA (HL)
Definition: A neoplastic proliferation of Reed-Sternberg (R-S) cells derived from germinal centre B cells, surrounded by a reactive inflammatory infiltrate.
Reed-Sternberg Cells:
- Large cells with abundant cytoplasm and multiple or bilobed nuclei ("owl-eye" appearance)
- Classic HL: CD15+ and CD30+ (B and T cell antigens usually negative)
- Lymphocyte-predominant HL (LPHL): CD20+, CD45+, CD15-
Clinical Features:
- Bimodal age distribution: 15-40 years (young adults) and >55 years
- Presents with painless peripheral adenopathy - most commonly cervical and supraclavicular
- B symptoms (in ~25% of patients):
- Fever >38°C (persistent)
- Drenching night sweats
- Unexplained weight loss >10% body weight in 6 months
- Severe generalised pruritus - more common in HL than NHL, often precedes diagnosis by months
- B symptoms + pruritus are due to cytokine production by R-S cells
Ann Arbor Staging (Cotswolds Modification):
| Stage | Description |
|---|---|
| I | Single lymph node region or lymphoid structure |
| II | Two or more regions on the same side of diaphragm |
| III | Lymph node regions on both sides of diaphragm |
| IV | Multiple extranodal sites or lymph nodes + extranodal disease |
- "B" suffix: B symptoms present
- "E" suffix: contiguous extranodal involvement
- "X": bulky disease (>1/3 mediastinal widening or >10 cm nodal mass)
Treatment:
- ABVD chemotherapy (Adriamycin, Bleomycin, Vinblastine, Dacarbazine) ± radiotherapy
- Excellent prognosis - one of the most curable malignancies
Cummings Otolaryngology Head & Neck Surgery
5. THALASSAEMIA
Definition: A group of hereditary anaemias caused by a defect in haemoglobin peptide chain synthesis, transmitted as an autosomal recessive trait.
Types:
| Type | Defect |
|---|---|
| Beta-thalassaemia (most common) | Reduced beta-chain synthesis → reduced HbA |
| Alpha-thalassaemia | Reduced alpha-chain synthesis |
| Gamma-thalassaemia | Reduced gamma-chain synthesis |
Pathophysiology:
- Reduced chain synthesis → intracellular precipitates called Heinz bodies → premature RBC destruction
- Severity: Heterozygous = thalassaemia minor (mild/carrier); Homozygous = thalassaemia major (severe)
Thalassaemia Major (Cooley's Anaemia / Mediterranean Anaemia):
Clinical features appearing in the first year of life:
- Chronic anaemia, jaundice, splenomegaly
- Retarded growth
- Enlarged head with slanting eyes and depressed nose (due to extramedullary haematopoiesis)
- Leg ulcers, abdominal distension
Blood film:
- Microcytic, hypochromic red cells
- Small, thin, misshapen RBCs with resistance to osmotic lysis
- Nucleated RBCs + other immature blood cells in severe forms
Diagnosis: Confirmed by haemoglobin electrophoresis
Treatment:
- Blood transfusions (may become transfusion-dependent)
- Splenectomy: for hypersplenism requiring frequent transfusions or when haemolytic antibodies develop
- Iron chelation (desferrioxamine) to prevent transfusion iron overload
- Bone marrow transplant (curative)
Bailey and Love's Short Practice of Surgery; Grainger & Allison's Diagnostic Radiology
6. SICKLE CELL ANAEMIA
Definition: A hereditary, autosomal recessive haemolytic anaemia in which normal HbA is replaced by haemoglobin S (HbS).
Pathophysiology:
- Point mutation: Glutamic acid → Valine at position 6 of the beta-globin chain
- Under low oxygen tension: HbS crystallises → distorts and elongates the RBC into a sickle shape
- Consequences: increased blood viscosity → microvascular occlusion → ischaemia and infarction
Epidemiology:
- Mainly those of African origin
- Sickle cell trait (HbAS carrier): 9% of Africans - usually asymptomatic
- Sickle cell disease (HbSS): ~1% of Africans
Clinical Features (Vaso-occlusive Crises):
| System Affected | Manifestation |
|---|---|
| Bone/joints | Bone pain, dactylitis |
| CNS | Stroke, neurological abnormalities |
| Spleen | Splenic microinfarcts → autosplenectomy |
| Kidney | Haematuria, renal papillary necrosis |
| Penis | Priapism |
| Skin | Leg ulcers |
| Abdomen | Visceral blood stasis, pain |
Diagnosis:
- Blood film: sickle-shaped cells
- Haemoglobin electrophoresis (gold standard - has largely replaced blood film)
Treatment:
- Hydroxyurea (increases HbF production)
- Prophylactic penicillin + vaccinations (due to functional asplenia)
- Analgesia for crises; IV fluids, O₂
- Blood transfusion for severe crises
- Bone marrow transplant (curative)
Bailey and Love's Short Practice of Surgery
7. DISSEMINATED INTRAVASCULAR COAGULATION (DIC)
Definition: Widespread systemic activation of coagulation resulting in microvascular thrombosis throughout the body, with simultaneous consumption of clotting factors and platelets, and secondary activation of fibrinolysis.
Also called: Consumptive coagulopathy
Paradox of DIC:
- Excessive clotting AND bleeding coexist in the same patient
- More commonly causes bleeding than all congenital coagulation disorders combined
Pathogenesis (2 main triggers):
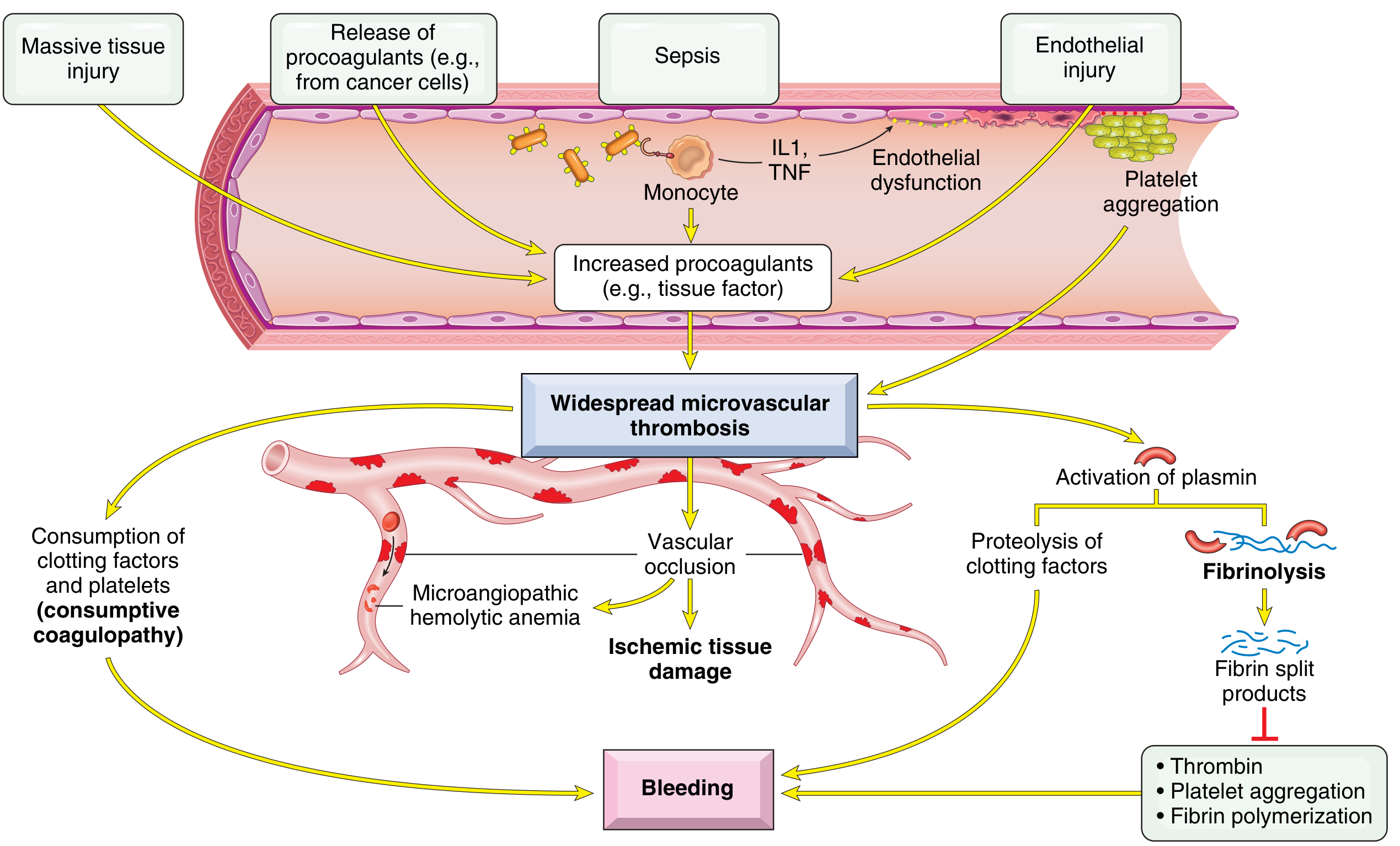
-
Release of tissue factor / procoagulants into circulation:
- Obstetric complications (amniotic fluid embolism, abruptio placentae)
- Cancer cells (especially acute promyelocytic leukaemia, adenocarcinoma)
- Bacterial sepsis (endotoxins stimulate tissue factor on monocytes → IL-1 + TNF → tissue factor on endothelium + downregulation of thrombomodulin)
-
Widespread endothelial injury:
- Antigen-antibody complexes (SLE)
- Temperature extremes (heat stroke, burns)
- Infections (meningococci, rickettsiae)
Result:
- Microvascular thrombi → ischaemic tissue damage
- Consumption of platelets + clotting factors → bleeding
- Secondary fibrinolysis → fibrin split products → inhibit thrombin, platelet aggregation, fibrin polymerization → further bleeding
- Microangiopathic haemolytic anaemia (RBCs sheared by fibrin strands)
Labs:
| Test | Result in DIC |
|---|---|
| PT / aPTT | Prolonged |
| Platelets | Low |
| Fibrinogen | Low |
| D-dimers / FDPs | High |
| Blood film | Schistocytes |
Causes (mnemonic - STOP Making New Thrombi):
- Sepsis (most common)
- Trauma / massive tissue injury
- Obstetric emergencies (abruption, eclampsia, amniotic fluid embolism)
- Promyelocytic leukaemia (M3 AML)
- Malignancy
- Necrotising pancreatitis
- Transfusion reactions
Robbins & Kumar Basic Pathology, p.427
8. BLOOD GROUPING (ABO System)
Basis: ABO antigens are carbohydrate structures linked to cell surface proteins and lipids, synthesised by polymorphic glycosyltransferase enzymes (gene on chromosome 9).
Molecular Basis:
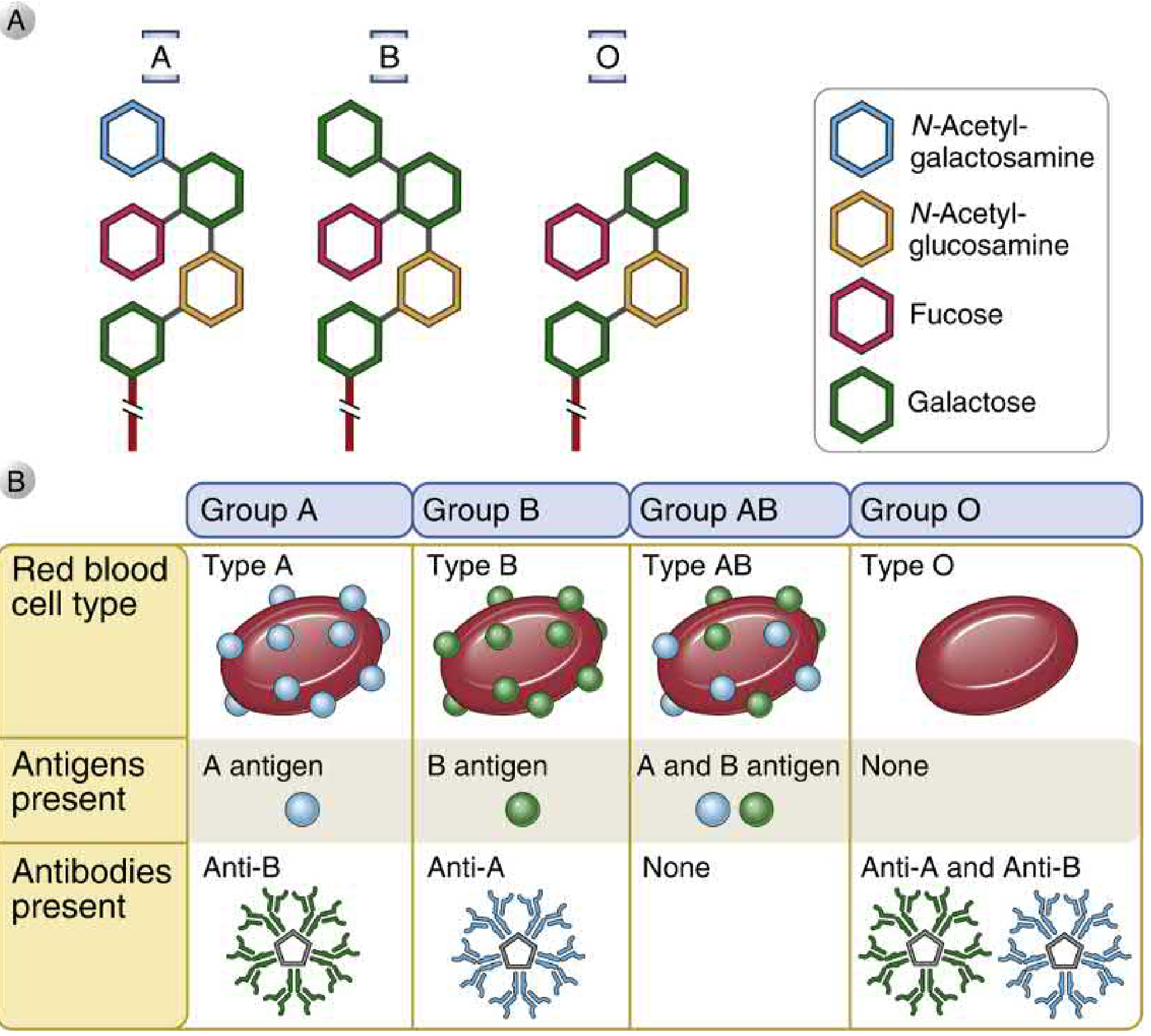
- All individuals produce a common core glycan
- H antigen: core glycan + fucose (added by fucosyltransferase) - the precursor
- A allele: adds terminal N-acetylgalactosamine to H antigen → A antigen
- B allele: adds terminal galactose to H antigen → B antigen
- O allele: no enzymatic activity → only H antigen expressed
Blood Group Summary:
| Blood Group | Genotype | Antigen on RBC | Antibody in Serum | Can Donate To | Can Receive From |
|---|---|---|---|---|---|
| A | AA or AO | A | Anti-B | A, AB | A, O |
| B | BB or BO | B | Anti-A | B, AB | B, O |
| AB | AB | A and B | None | AB only | ALL (universal recipient) |
| O | OO | None (H only) | Anti-A and Anti-B | ALL (universal donor) | O only |
Why Do We Have Natural Antibodies?
- Anti-A and Anti-B antibodies form against glycolipids of intestinal bacteria that cross-react with ABO antigens
- Individuals are tolerant to their own antigens - they only make antibodies against the antigens they lack
Special Note - Bombay Blood Group (hh):
- Rare mutation in fucosyltransferase gene → cannot produce H antigen
- Result: cannot produce H, A, or B antigens
- Makes antibodies against H, A, and B - incompatible with all standard blood groups
Rh System:
- D antigen is the most clinically significant
- Rh+ = D antigen present; Rh- = absent
- Anti-D antibodies are immune (not naturally occurring) - form after exposure to Rh+ blood
- Critical in: haemolytic disease of the newborn (HDN/erythroblastosis fetalis)
Cellular and Molecular Immunology; Histology: A Text and Atlas; Emery's Elements of Medical Genetics and Genomics
Quick Comparison Table
| Disease | Key Defect | Key Lab Finding | Key Treatment |
|---|---|---|---|
| Leukaemia | Clonal WBC proliferation | Blasts on blood film/bone marrow | Chemotherapy |
| Anaemia | Low Hb/RBC | Low Hb, altered MCV | Treat cause (iron, B12, transfusion) |
| ITP (Purpura) | Antiplatelet IgG | Low platelets, normal PT/aPTT | Steroids, IVIG, splenectomy |
| Hodgkin Lymphoma | R-S cells (B-cell neoplasm) | CD15+, CD30+, Reed-Sternberg cells | ABVD chemo ± radiotherapy |
| Thalassaemia | Defective Hb chain synthesis | Low MCV, Heinz bodies, HbF↑ on electrophoresis | Transfusion, chelation, BMT |
| Sickle Cell Anaemia | HbS polymerisation | Sickle cells on film, HbS on electrophoresis | Hydroxyurea, analgesia, BMT |
| DIC | Systemic coagulation activation | ↑PT, ↑aPTT, ↓platelets, ↓fibrinogen, ↑D-dimers | Treat cause, FFP, platelets |
| Blood Grouping | ABO carbohydrate antigens | ABO + Rh typing | Cross-match before transfusion |
All content sourced from authoritative medical textbooks including Robbins & Kumar Basic Pathology, Harrison's Principles of Internal Medicine 22E, Bailey and Love's Short Practice of Surgery, Cummings Otolaryngology, and Cellular and Molecular Immunology. Good luck studying! 📖
Teach me everything simply
Here's everything explained simply - like a story, not a textbook. 🩸
🩸 Blood Disorders - Simply Explained
1. 🔴 ANAEMIA - "Not Enough Good Blood"
Think of it like this: Your blood is like a delivery truck for oxygen. Anaemia means you don't have enough trucks - or the trucks are broken.
Why does it happen?
- Not making enough RBCs - lack of iron, B12, folate, or bone marrow problems
- Destroying RBCs too fast - sickle cell, thalassaemia, immune attack
- Losing blood - bleeding from ulcer, heavy periods, injury
Symptoms (all make sense when you understand "less oxygen delivery"):
- Tired all the time 😴
- Pale skin, pale inner eyelids
- Breathless on exertion
- Fast heartbeat
- Headache, dizziness
Quick Test:
- Check Haemoglobin (Hb) - low = anaemia
- Check MCV (size of RBCs):
- Small RBCs = iron deficiency or thalassaemia
- Normal RBCs = bleeding or chronic disease
- Big RBCs = B12/folate deficiency
2. ⚪ LEUKAEMIA - "Blood Cancer"
Think of it like this: Your bone marrow is a factory. Normally it makes good blood cells. In leukaemia, one cell goes rogue and starts making millions of useless, cancerous copies of itself - they fill the factory and crowd out the good workers.
Types - easy way to remember:
| Acute (fast, aggressive) | Chronic (slow, sneaky) | |
|---|---|---|
| Myeloid | AML | CML |
| Lymphoid | ALL | CLL |
- Acute = immature cells (blasts) - more dangerous, needs urgent treatment
- Chronic = more mature cells - slower progression
Why do patients get sick?
Because normal cells get crowded out:
- ❌ Not enough RBCs → anaemia (tired, pale)
- ❌ Not enough platelets → bleeding, bruising
- ❌ Not enough normal WBCs → recurrent infections
- ✅ Too many useless white cells → swollen lymph nodes, enlarged liver/spleen
Diagnosis: Blood film + Bone marrow biopsy
Treatment: Chemotherapy (targeted therapy for CML - Imatinib/Gleevec)
3. 🟣 PURPURA - "Blood Spots Under the Skin"
Think of it like this: Tiny blood vessels are leaking blood into the skin. The spots are red/purple and - KEY POINT - they don't go white when you press them (this is how you tell it's blood, not just redness).
Two main types:
Non-palpable (flat spots):
- Small spots = petechiae
- Larger patches = ecchymosis (bruise)
- Cause: too few platelets (thrombocytopenia) or clotting problem
Palpable (raised spots):
- You can feel them
- Cause: vasculitis (inflamed blood vessels)
Most Important Cause - ITP (Immune Thrombocytopenic Purpura):
- Your immune system makes antibodies that attack your OWN platelets
- Kids: after a viral infection → usually gets better on its own
- Adults (usually young women): becomes chronic (>6 months)
- Platelets are low, but PT and aPTT are normal (clotting factors are fine)
- Treatment: Steroids first → IVIG → Splenectomy (spleen is where platelets are being destroyed)
Rule to remember:
Purpura = bleeding into skin = does NOT blanch on pressure ✅ Red rash from inflammation = DOES blanch on pressure ❌
4. 🎗️ HODGKIN LYMPHOMA - "Cancer of Lymph Nodes with an Owl-Eye Cell"
Think of it like this: Lymph nodes are your body's "security checkpoints." In Hodgkin Lymphoma, one type of B-cell goes cancerous and creates a huge inflammatory party in the lymph nodes.
The STAR of this disease - Reed-Sternberg Cell:
- Huge cell with two big nuclei that look like owl eyes 🦉
- This is the diagnostic cell - if you see it in a biopsy, it's Hodgkin
- It releases cytokines that cause most of the symptoms
Who gets it?
- Young adults (15-40 years) - or elderly (>55)
- Starts in the neck/supraclavicular lymph nodes
Classic Symptoms:
- Painless swollen lymph nodes (usually neck)
- B symptoms:
- Fever >38°C
- Drenching night sweats
- Weight loss >10% in 6 months
- Itch (pruritus) - often the first clue, months before diagnosis
- Pain in lymph nodes after drinking alcohol (unique to Hodgkin!)
Staging - Ann Arbor System:
Think of the body divided by the diaphragm (breathing muscle in the middle):
- Stage I = one area
- Stage II = two areas, same side of diaphragm
- Stage III = both sides of diaphragm
- Stage IV = organs involved (liver, bone marrow, lungs)
- Add "B" if B symptoms present
Treatment: ABVD chemotherapy ± radiotherapy
Prognosis: EXCELLENT - one of the most curable cancers 🎉
5. 🧬 THALASSAEMIA - "Broken Haemoglobin Factory"
Think of it like this: Haemoglobin (what carries oxygen in RBCs) is made of chains. Thalassaemia means you can't make enough of one of those chains. It's like trying to make a pair of scissors but only having one blade.
The chains of Haemoglobin:
- Normal adult Hb (HbA) = 2 alpha chains + 2 beta chains
- Beta-thalassaemia = can't make enough beta chains (most common)
- Alpha-thalassaemia = can't make enough alpha chains
Severity:
| Thalassaemia Minor | Thalassaemia Major | |
|---|---|---|
| Genes | One gene broken (carrier) | Both genes broken |
| Symptoms | Mild anaemia, mostly fine | Severe disease from infancy |
Thalassaemia Major - what happens:
- Severe anaemia from first year of life
- The bone marrow works overtime trying to make RBCs → bones expand → characteristic facial features:
- Large head
- Prominent cheekbones
- Slanting eyes, depressed nose bridge
- "Chipmunk facies"
- Jaundice (from RBC destruction)
- Massive splenomegaly (spleen trying to clear broken RBCs)
- Growth retardation
- Leg ulcers
The broken RBCs:
- Unmatched chains form Heinz bodies (clumps inside RBC) → premature destruction
- RBCs are small, thin, misshapen
- Resistant to osmotic lysis (important lab finding)
Diagnosis: Haemoglobin electrophoresis (shows HbF↑, HbA2↑, HbA↓)
Treatment:
- Regular blood transfusions (but patient can become dependent)
- Iron chelation (desferrioxamine) - transfusions load the body with iron
- Splenectomy if hypersplenism or haemolytic antibodies develop
- Bone marrow transplant = only cure
6. 🌙 SICKLE CELL ANAEMIA - "Blood Cells That Turn Into Sickles"
Think of it like this: Normal RBCs are like round, flexible frisbees that flow easily through blood vessels. In sickle cell, when oxygen drops, they turn into rigid, crescent/sickle shapes that get stuck and block blood flow - like traffic jams in tiny blood vessels.
The Mutation (dead simple):
- One letter change in DNA
- Glutamic acid → Valine at position 6 of beta-globin chain
- This makes HbS instead of HbA
- Under low O₂: HbS crystallises → RBC becomes a sickle
Who gets it?
- Mainly people of African origin
- Trait (HbAS): one copy - usually no symptoms, slight protection against malaria
- Disease (HbSS): two copies - full disease
What goes wrong (Vaso-occlusive Crisis):
The sickled cells block small blood vessels → ischaemia and pain wherever the blockage is:
| Where Blocked | What Happens |
|---|---|
| Bones | Severe bone pain (most common crisis) |
| Hands/feet in babies | Dactylitis (sausage fingers) |
| Spleen | Repeated infarcts → autosplenectomy (spleen destroys itself) |
| Brain | Stroke |
| Penis | Priapism (painful prolonged erection) |
| Kidneys | Haematuria, renal failure |
| Skin | Leg ulcers |
Key Complication: Autosplenectomy
- Repeated splenic infarcts destroy the spleen
- Patient becomes functionally asplenic → vulnerable to encapsulated bacteria (Pneumococcus, Meningococcus, Haemophilus)
- These patients MUST be on prophylactic penicillin + vaccines
Diagnosis: Sickle cells on blood film → confirmed by haemoglobin electrophoresis
Treatment:
- Hydroxyurea - increases HbF (fetal haemoglobin) which doesn't sickle - best medical treatment
- Analgesia + IV fluids + O₂ for crises
- Prophylactic penicillin + vaccinations
- BMT = only cure
7. ⚡ DIC - "The Body That's Trying to Clot AND Bleed at the Same Time"
Think of it like this: Imagine your fire alarm system goes haywire and sets off sprinklers everywhere in the building - both inside AND outside the rooms that are on fire. That's DIC - the clotting system gets turned on everywhere at once.
What is it?
- Normally, clotting is a local, controlled process
- In DIC, something triggers whole-body clotting activation at once
- All the clotting factors and platelets get used up
- Then when you actually need to clot, you CAN'T → you BLEED
The Paradox:
Clotting + Bleeding happening at the SAME time in the same patient
What triggers it?
- Sepsis (most common - bacteria release endotoxins)
- Obstetric emergencies (amniotic fluid embolism, placental abruption, eclampsia)
- Trauma / burns (tissue factor release)
- Acute Promyelocytic Leukaemia (M3-AML - cancer cells release procoagulants)
- Cancer (mucin-secreting adenocarcinomas)
- Transfusion reactions
What happens step by step:
- Trigger activates tissue factor everywhere
- Widespread microvascular clots form → organs get ischaemic damage
- Clotting factors + platelets all get consumed
- Fibrinolysis (clot-busting) gets activated → fibrin split products form
- These split products inhibit further clotting → massive BLEEDING
- RBCs get chopped up passing through fibrin strands → microangiopathic haemolytic anaemia (schistocytes on blood film)
Lab Findings (everything is deranged):
| Test | Result |
|---|---|
| PT & aPTT | ↑ PROLONGED |
| Platelets | ↓ LOW |
| Fibrinogen | ↓ LOW |
| D-dimers | ↑ HIGH |
| Blood film | Schistocytes (fragmented RBCs) |
Treatment:
- Treat the underlying cause (most important!)
- FFP (fresh frozen plasma) - replaces clotting factors
- Platelet transfusion
- Cryoprecipitate (for fibrinogen)
- Heparin in some cases (controversial)
8. 🅰️🅱️ BLOOD GROUPING - "Your Blood's ID Card"
Think of it like this: Your red blood cells carry a "flag" on their surface. Your blood group is just which flag you carry. Your immune system is trained to attack foreign flags.
ABO System - The 4 Flags:
| Group | Flag on RBC | Antibody in Blood | Donate to | Receive from |
|---|---|---|---|---|
| A | A antigen | Anti-B | A, AB | A, O |
| B | B antigen | Anti-A | B, AB | B, O |
| AB | A + B antigen | None | AB only | ALL ← universal recipient |
| O | No antigen | Anti-A + Anti-B | ALL ← universal donor | O only |
How the flags are made (simple chemistry):
- Everyone starts with a base "H antigen"
- Gene A adds a sugar → makes A antigen
- Gene B adds a different sugar → makes B antigen
- Gene O does nothing → only H antigen remains
Why do you have antibodies against flags you've never been transfused with?
- Because gut bacteria have similar sugars on their surface
- Your immune system accidentally makes antibodies against the ABO antigens it doesn't have (via cross-reaction with bacteria)
- So Anti-A and Anti-B are naturally occurring - you have them from birth
Bombay Blood Group (hh) - Super Rare:
- Can't even make the H antigen (the base)
- So can't make A, B, or O antigens
- Makes anti-H, anti-A, and anti-B antibodies
- Incompatible with ALL normal blood groups
- Must receive blood from other Bombay donors only
Rh System:
- D antigen = most important after ABO
- Rh+ (most people) = have D antigen
- Rh- = no D antigen
- Danger: Rh- mother carrying Rh+ baby
- During delivery, baby's blood enters mother's circulation
- Mother makes Anti-D antibodies
- In next pregnancy, Anti-D crosses placenta → attacks baby's RBCs → Haemolytic Disease of the Newborn (HDN)
- Prevented by giving Anti-D immunoglobulin to Rh- mothers
Before Transfusion - 3 Steps:
- ABO + Rh typing (which group is the patient?)
- Antibody screen (does patient have unexpected antibodies?)
- Cross-match (mix donor RBCs with patient serum - no reaction = safe to transfuse)
🧠 The Big Picture - How They All Connect
BLOOD DISORDERS
├── Too FEW RBCs → ANAEMIA
│ ├── Production problem → Iron deficiency, B12, thalassaemia
│ ├── Destruction → Sickle cell, thalassaemia, DIC
│ └── Blood loss → Haemorrhage
│
├── WRONG RBCs → SICKLE CELL & THALASSAEMIA
│ ├── Wrong shape (sickle) → blocks vessels → pain crises
│ └── Wrong chains (thalassaemia) → fragile cells → haemolysis
│
├── White cell CANCER → LEUKAEMIA
│ └── Crowds out good cells → anaemia + bleeding + infections
│
├── Lymph node CANCER → HODGKIN LYMPHOMA
│ └── Reed-Sternberg cells, B symptoms, owl-eye nuclei
│
├── BLEEDING into skin → PURPURA
│ └── Low platelets (ITP) or vasculitis
│
└── System-wide CLOTTING CHAOS → DIC
└── Triggered by sepsis/trauma/cancer
└── Clot everywhere + bleed everywhere
🔑 The 10 Most Exam-Important Facts
- Anaemia - MCV tells you the cause (small=iron/thal, big=B12/folate)
- Leukaemia - blasts on bone marrow; acute=more aggressive
- Purpura = does NOT blanch on pressure
- ITP - low platelets only; PT/aPTT normal; treat with steroids first
- Hodgkin Lymphoma - Reed-Sternberg cells (CD15+, CD30+); B symptoms; excellent prognosis
- Thalassaemia major - chipmunk facies + splenomegaly + Heinz bodies; diagnosed by Hb electrophoresis
- Sickle cell - HbS crystallises under low O₂; autosplenectomy → needs penicillin prophylaxis; hydroxyurea is treatment
- DIC - consumptive coagulopathy; ↑PT, ↑aPTT, ↓platelets, ↓fibrinogen, ↑D-dimers, schistocytes
- Blood group O = universal donor; AB = universal recipient
- Bombay group = can't make H antigen; incompatible with all normal blood
Study these stories, not just the facts - and the exam answers will come naturally! 💪
This is a shared conversation. Sign in to Orris to start your own chat.