Glucocorticoids
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
I now have all the information needed for a thorough, well-illustrated answer. Let me compose it.
Glucocorticoids
Overview
Glucocorticoids are steroid hormones produced by the zona fasciculata of the adrenal cortex. Cortisol (hydrocortisone) is the principal endogenous glucocorticoid. Synthetic analogs - prednisone, dexamethasone, methylprednisolone, and others - are among the most widely used drugs in medicine, with applications ranging from replacement therapy to anti-inflammatory and immunosuppressive treatment.
The term "corticosteroid" is often used synonymously with glucocorticoid, but the preferred designation when using exogenous agents therapeutically is "glucocorticoid," since corticosteroid encompasses both glucocorticoid and mineralocorticoid hormones. (Rheumatology, 2-Volume Set)
Endogenous Physiology
Cortisol is released under the control of the hypothalamic-pituitary-adrenal (HPA) axis:
- Hypothalamus releases corticotropin-releasing hormone (CRH)
- Anterior pituitary releases ACTH
- Adrenal cortex releases cortisol, which feeds back to suppress both CRH and ACTH
Cortisol shows a diurnal rhythm: peak levels around 7-8 AM and a nadir around midnight. Endogenous daily production averages approximately 5.7 mg/m²/day of hydrocortisone. (Rheumatology, 2-Volume Set)
Structure-Activity Relationships
The 11β and 17α hydroxyl groups are critical for glucocorticoid activity. Synthetic analogs are built by modifying the cortisol scaffold:
- Adding a double bond in ring A (prednisone/prednisolone) enhances glucocorticoid activity without increasing mineralocorticoid activity
- 6α methylation produces methylprednisolone (further increases glucocorticoid activity)
- 9α fluorination yields triamcinolone
- Both 6α methylation and 9α fluorination together yield dexamethasone
Prednisone is a prodrug - it is rapidly converted to the active form prednisolone in the liver by CYP3A4. Prednisolone is preferred in patients with significant hepatic dysfunction. (Rheumatology, 2-Volume Set)
Potency Comparison
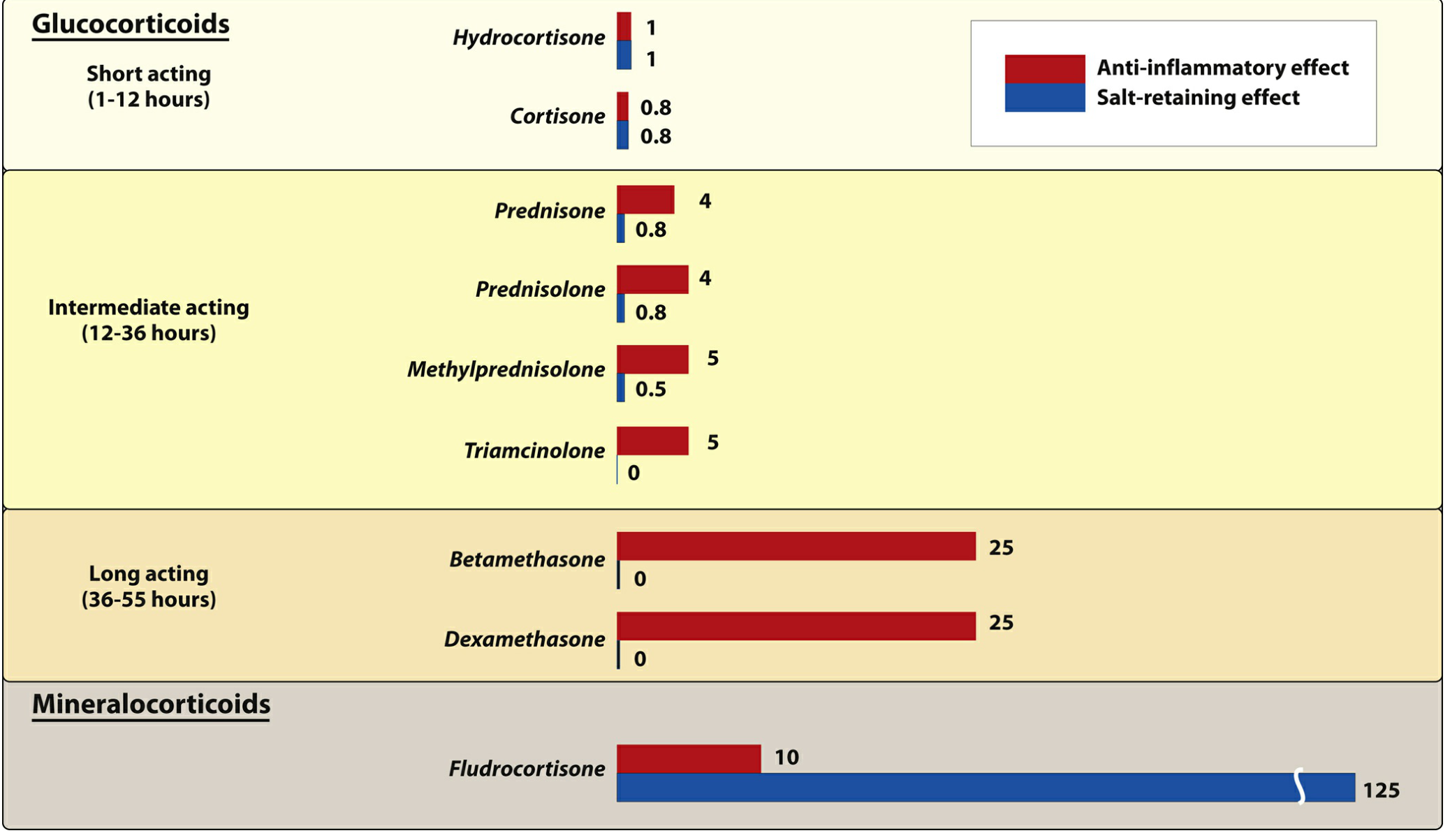
Figure: Pharmacologic effects and duration of action of commonly used natural and synthetic corticosteroids (relative to hydrocortisone = 1). (Lippincott Illustrated Reviews: Pharmacology)
| Glucocorticoid | Equivalent Oral Dose (mg) | Plasma t½ (min) | Anti-inflammatory Potency | Mineralocorticoid Potency |
|---|---|---|---|---|
| Cortisol (hydrocortisone) | 20 | 90 | 1 | 1 |
| Prednisone / Prednisolone | 5 | 200 | 4 | 0.8 |
| Methylprednisolone | 4 | 200 | 5 | 0.5 |
| Triamcinolone | 4 | 200 | 5 | 0 |
| Dexamethasone | 0.75 | 300 | 25 | 0 |
(Rheumatology, 2-Volume Set, Table 62.1)
Mechanism of Action
Glucocorticoids act through genomic and nongenomic pathways:
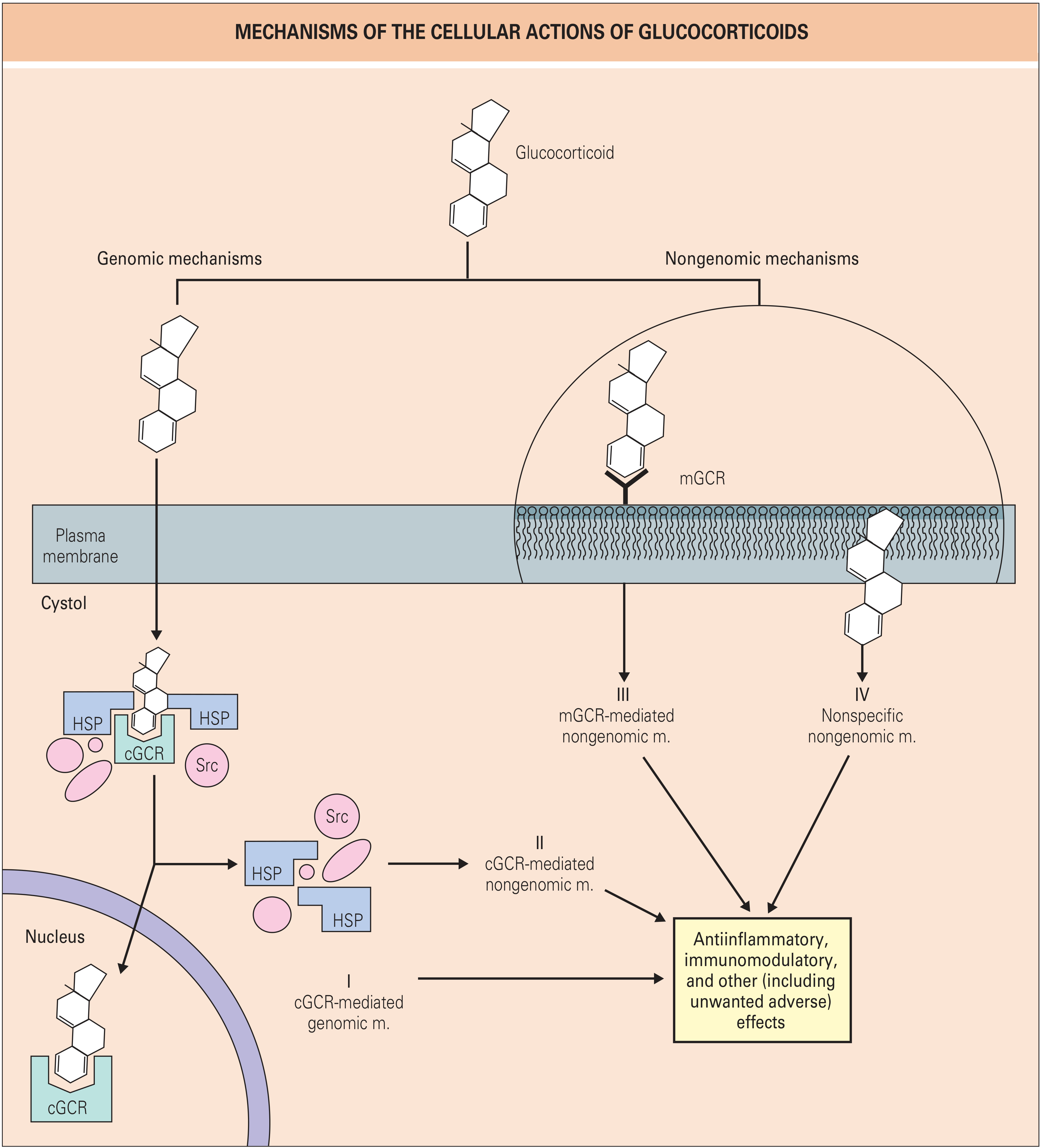
Figure: Mechanisms of the cellular actions of glucocorticoids. Four distinct pathways (I-IV) lead to anti-inflammatory, immunomodulatory, and other effects. (Rheumatology, 2-Volume Set)
Genomic Pathways
-
cGCR-mediated genomic mechanism (Pathway I): Glucocorticoids are lipophilic and freely cross the cell membrane. In the cytosol, they bind to the cytosolic glucocorticoid receptor (cGCR), which is held in an inactive state by heat shock proteins (HSP90, HSP70) and Src kinase. Ligand binding causes dissociation of the chaperone complex. The glucocorticoid-receptor complex is chaperoned to the nucleus, where it binds to glucocorticoid response elements (GREs) in gene promoters or suppressors to activate or repress transcription.
-
Transrepression via NF-κB and AP-1: The glucocorticoid-receptor complex directly inhibits key pro-inflammatory transcription factors - NF-κB and AP-1 - thereby suppressing transcription of genes encoding cytokines (IL-1, IL-6, TNF-α), COX-2, iNOS, and adhesion molecules. This is considered a primary anti-inflammatory mechanism.
-
cGCR-mediated nongenomic mechanism (Pathway II): Released Src kinase from the HSP complex triggers rapid cytoplasmic signaling independently of gene transcription.
Nongenomic Pathways
- Membrane glucocorticoid receptor (mGCR, Pathway III): Glucocorticoids bind membrane-associated receptors, producing rapid effects within seconds to minutes.
- Nonspecific membrane effects (Pathway IV): High-dose glucocorticoids intercalate into cell membranes, altering membrane biophysics. This is the likely basis for the pronounced nongenomic effects of IV pulse methylprednisolone (which has >3-fold more nongenomic effects than prednisolone at equivalent doses).
(Rheumatology, 2-Volume Set)
Anti-Inflammatory and Immunological Effects
Glucocorticoids produce broad suppression of the inflammatory response:
- Inhibit pro-inflammatory cytokines: IL-1β, IL-2, IL-6, IL-8, TNF-α, IFN-γ
- Inhibit pro-inflammatory enzymes: COX-2, phospholipase A2 (via lipocortin-1/annexin A1 induction), collagenase, elastase, plasminogen activator
- Decrease T-cell function and reduce circulating T-cell numbers
- Inhibit Fc receptor expression, reducing clearance of antibody-coated blood cells
- Increase circulating neutrophils (demargination from vessel walls) while impairing their migration to sites of inflammation
- Inhibit leukocyte adhesion to endothelial cells
The transcriptional response to glucocorticoids is highly cell-type specific - the individual genes and pathways affected, as well as the magnitude and direction of regulation, differ between cell types. (Rheumatology, 2-Volume Set)
Dosing Concepts
EULAR definitions for oral prednisone equivalent dosing:
- Low dose: ≤7.5 mg/day
- Medium dose: >7.5 to ≤30 mg/day
- High dose: >30 to ≤100 mg/day
- Very high / pulse therapy: 250-1000 mg/day IV methylprednisolone for 3 days (for life- or organ-threatening disease)
Timing matters: Prednisolone and methylprednisolone clearance is up to 25% lower in the morning than in the evening. Modified-release prednisone (taken at night, releasing 4 hours later) reduces morning stiffness in RA more effectively than immediate-release forms. (Rheumatology, 2-Volume Set)
Therapeutic Uses
| Indication | Notes |
|---|---|
| Replacement therapy (Addison's disease) | Hydrocortisone mimics diurnal cortisol; 2/3 dose in morning, 1/3 afternoon |
| Rheumatoid arthritis | Low-dose reduces disease activity and slows radiographic progression |
| Polymyalgia rheumatica / Giant cell arteritis | Mainstay of therapy |
| SLE, inflammatory myopathy, systemic vasculitis | Usually 1 mg/kg/day; pulse therapy for severe CNS/renal involvement |
| Asthma / allergic rhinitis | Inhaled steroids (fluticasone, budesonide) for long-term control |
| Organ transplant rejection prophylaxis | Immunosuppressive effect |
| Cerebral edema | Dexamethasone (no mineralocorticoid activity) |
| Cushing syndrome diagnosis | Dexamethasone suppression test |
| Septic shock (select cases), ARDS | Hydrocortisone ± fludrocortisone |
| Intra-articular injection | Osteoarthritis flares |
(Lippincott Illustrated Reviews: Pharmacology; Rheumatology, 2-Volume Set)
Adverse Effects
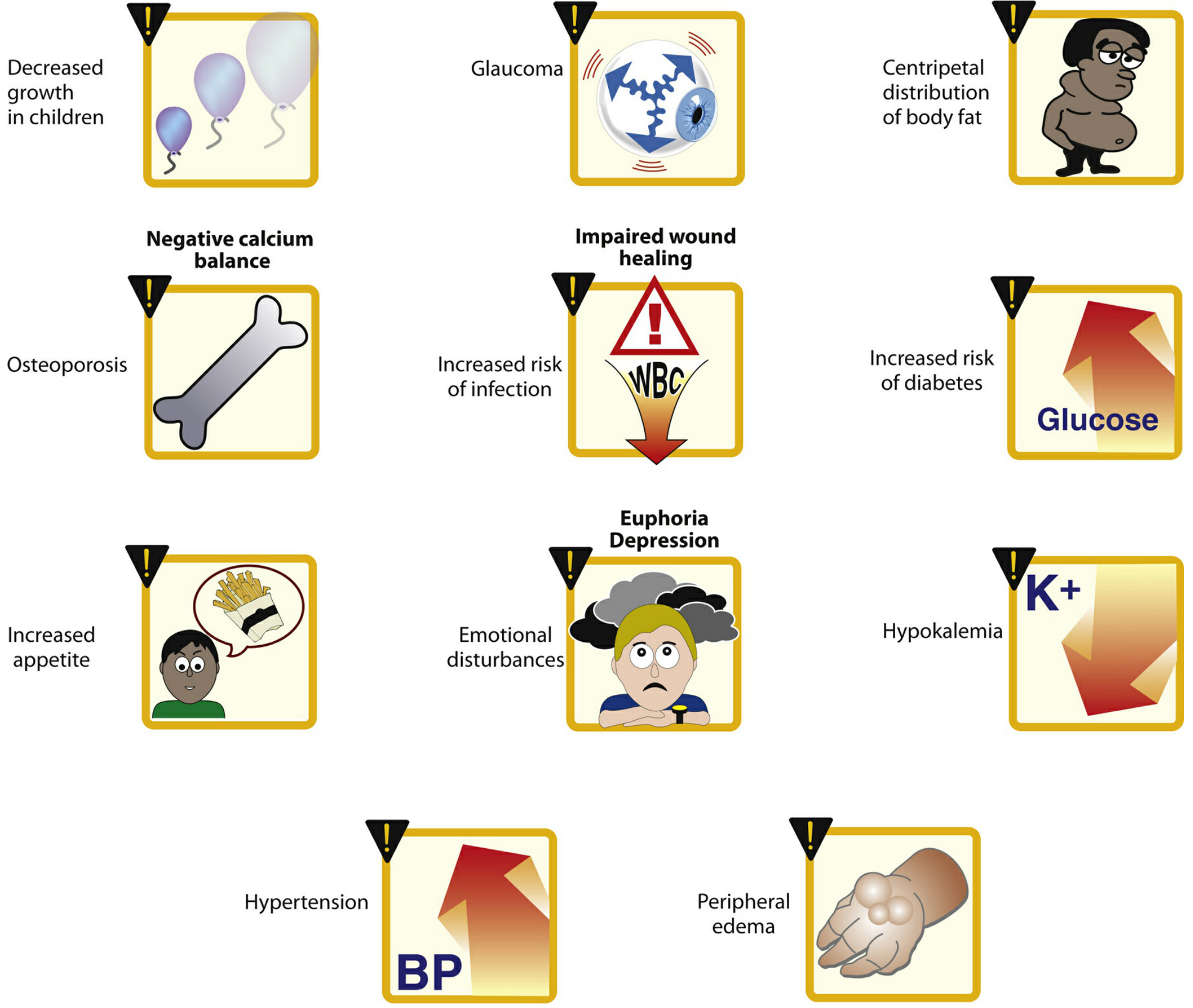
Figure: Commonly observed effects of long-term corticosteroid therapy. (Lippincott Illustrated Reviews: Pharmacology)
Key Adverse Effects (dose-dependent)
Musculoskeletal
- Glucocorticoid-induced osteoporosis (GIOP): The most devastating long-term complication. Up to 40% of users develop bone loss leading to fracture. Predominantly affects trabecular bone initially (vertebrae), later cortical bone (femoral neck). First-year bone loss can be 1.5-20% at ≤10 mg/day. Risk mitigation requires calcium/vitamin D supplementation and bisphosphonate therapy.
- Proximal myopathy: Muscle weakness, particularly in the thighs and shoulder girdle
Metabolic
- Hyperglycemia / steroid-induced diabetes mellitus
- Central (centripetal) obesity, moon facies, buffalo hump (Cushing-like features)
- Hypertriglyceridemia
- Negative calcium balance
Cardiovascular
- Hypertension (sodium and water retention)
- Peripheral edema
Immunological
- Increased susceptibility to infection (bacterial, fungal, viral)
- Impaired wound healing
Endocrine
- HPA axis suppression (adrenal suppression) - risk of adrenal crisis on withdrawal
- Diminished sex hormone production
Neuropsychiatric
- Euphoria, emotional disturbances, depression, psychosis
- Insomnia
Ocular
- Posterior subcapsular cataracts
- Glaucoma (raised intraocular pressure)
Pediatric
- Growth retardation (mechanism unknown, partly via growth hormone suppression; alternate-day dosing reduces this effect)
Skin (topical)
- Skin atrophy, ecchymosis, striae, telangiectasia
(Rheumatology, 2-Volume Set; Lippincott Illustrated Reviews: Pharmacology)
Discontinuation and Adrenal Suppression
Abrupt discontinuation after prolonged therapy can precipitate acute adrenal insufficiency (adrenal crisis), which may be fatal. Manifestations include nausea, vomiting, fever, dehydration, hypotension, shock, hypoglycemia, and hyperkalemia.
Key rules:
- Doses must be tapered slowly according to individual tolerance
- Risk is present any time the HPA axis has been suppressed (typically courses >3 weeks or doses above physiologic replacement)
- Patients on chronic steroids may need stress dosing (increased dose) during surgery or serious illness
Drug Interactions
- Phenobarbital, phenytoin, rifampin: Induce CYP3A4, increasing glucocorticoid metabolism and reducing efficacy
- Concurrent aspirin: Accelerates salicylate metabolism, reducing salicylate levels
- NSAIDs: Additive GI risk (peptic ulceration)
- Antidiabetic drugs: Glucocorticoids antagonize their effects, requiring dose adjustments
Special Considerations
Glucocorticoid resistance: Reported in severe RA, associated with higher glucocorticoid receptor β levels, MAPK activation, and antibodies to lipocortin-1. The 11β-hydroxysteroid dehydrogenase isoform balance also plays a central role.
Selective GR agonists (SEGRAs): Under development to dissociate transrepression (anti-inflammatory) from transactivation (adverse metabolic effects). Fosdagrocorat in phase IIb trials showed similar efficacy to prednisone 10 mg/day with metabolic safety closer to prednisone 5 mg/day, with reduced HbA1c elevations.
Liposomal glucocorticoids: In development to provide prolonged drug concentrations at the site of inflammation with lower systemic exposure. (Rheumatology, 2-Volume Set)
Glucocorticoids Physiology
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive physiological content from Medical Physiology (Boron & Boulpaep) and Costanzo's Physiology. I have enough to write an outstanding, well-illustrated answer.
Glucocorticoids - Physiology
1. The Adrenal Gland: Source of Glucocorticoids
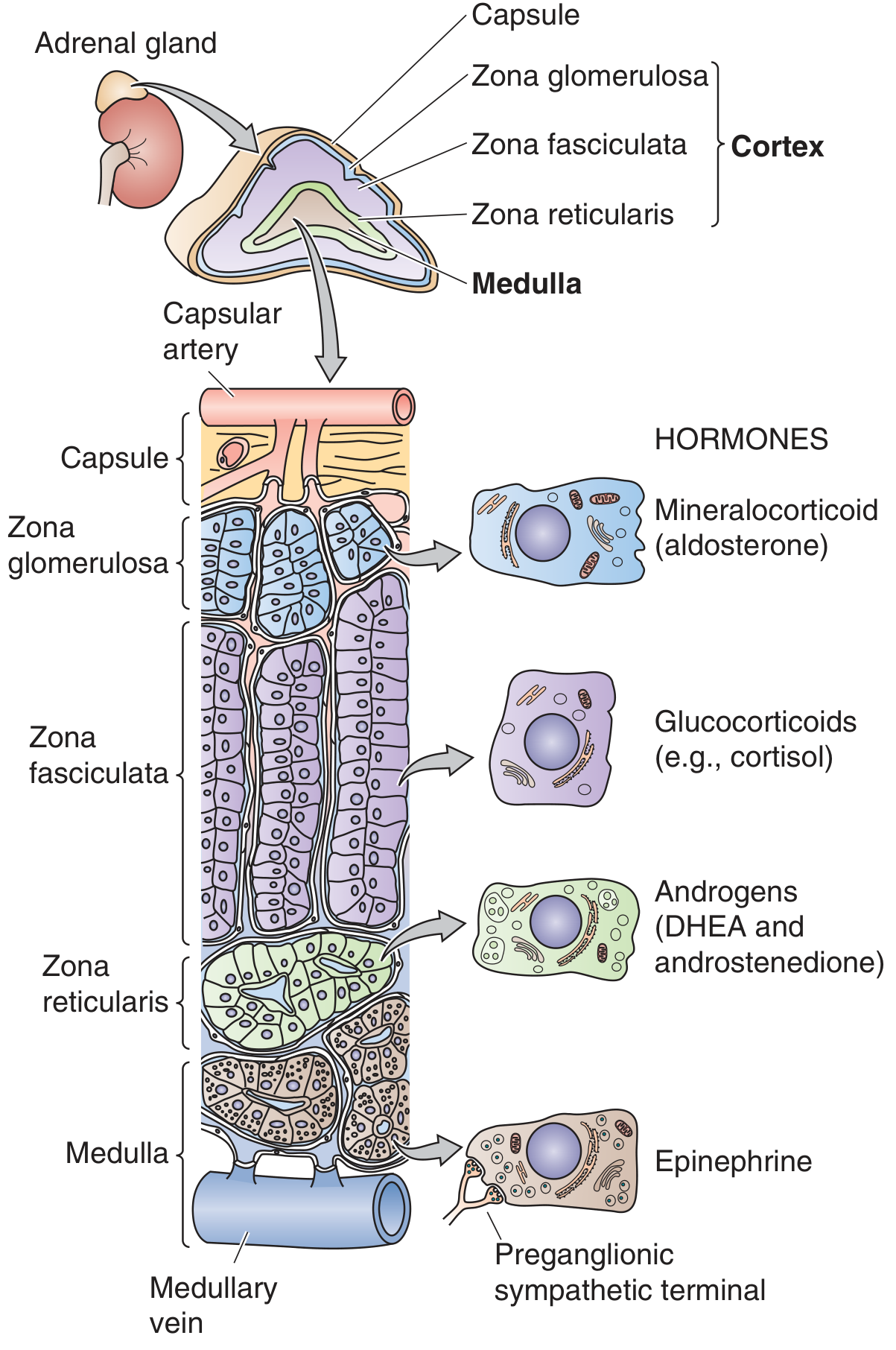
Figure: Anatomy of the adrenal gland. Each gland consists of an outer cortex (3 zones) and an inner medulla. The blood supply flows from the subcapsular region through the cortex into the medulla. (Medical Physiology, Boron & Boulpaep)
The adrenal cortex has three distinct zones, each producing different steroids:
| Zone | Product |
|---|---|
| Zona glomerulosa (outermost) | Mineralocorticoids (aldosterone) |
| Zona fasciculata (middle, largest) | Glucocorticoids (cortisol) |
| Zona reticularis (inner) | Adrenal androgens (DHEA, androstenedione) |
| Medulla | Catecholamines (epinephrine) |
Cortisol is the primary endogenous glucocorticoid in humans (corticosterone is the major glucocorticoid in rodents). Most body tissues are targets for glucocorticoid action - liver, fat, muscle, bone, skin, hematopoietic/lymphoid tissue, and the CNS all express glucocorticoid receptors. (Medical Physiology)
2. Biosynthesis: From Cholesterol to Cortisol
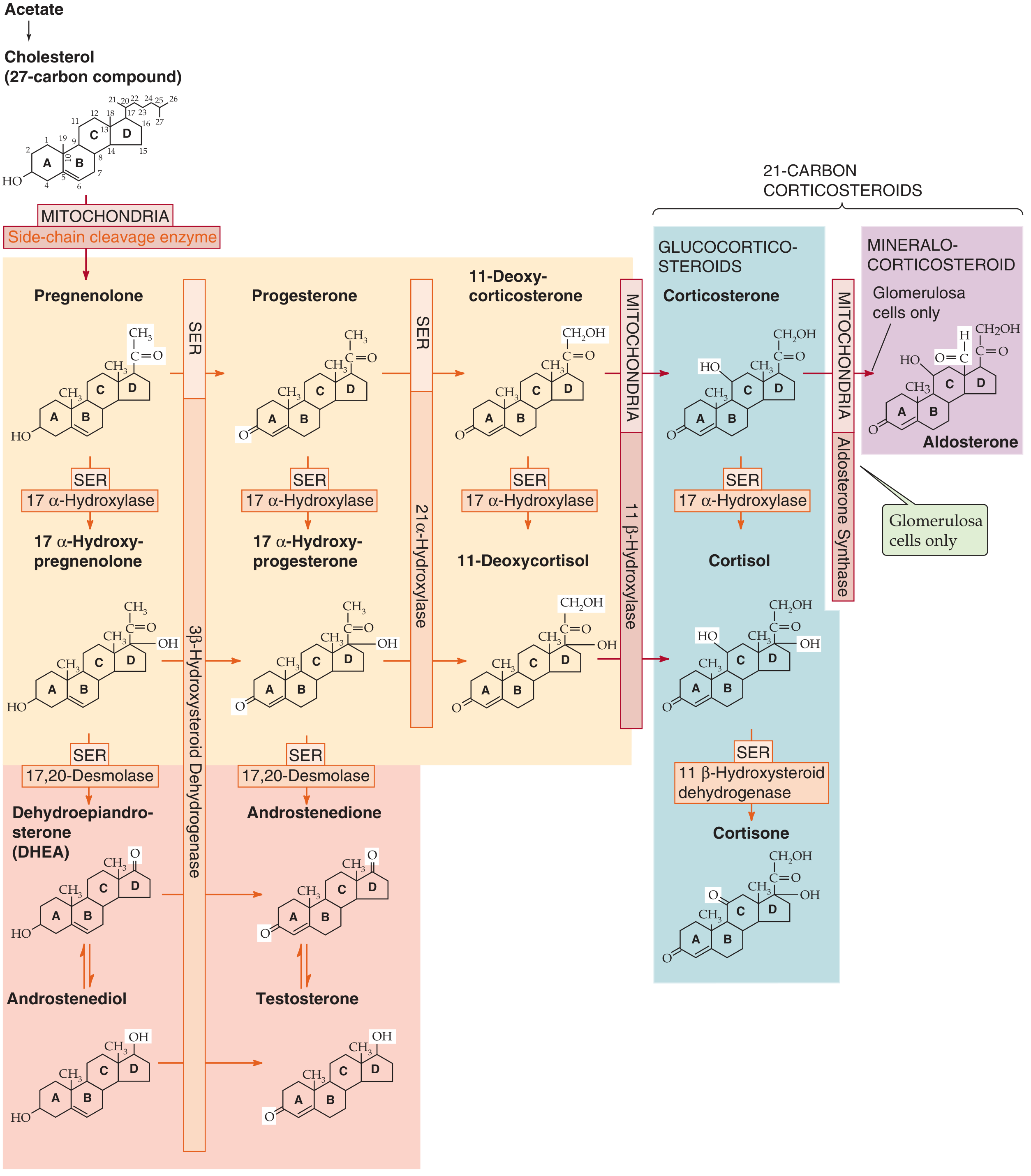
Figure: Biosynthesis of adrenal steroids. All steroids derive from cholesterol via a series of cytochrome P-450 enzyme reactions in the SER and mitochondria. (Medical Physiology)
Cortisol synthesis begins with cholesterol from two sources:
- Circulating LDL taken up by receptor-mediated endocytosis (quantitatively dominant)
- De novo synthesis from acetate
Five key enzymatic steps (zona fasciculata):
-
Cholesterol → Pregnenolone (mitochondria): Side-chain cleavage enzyme (P-450scc / 20,22-desmolase) removes carbons 22-27. This is the rate-limiting step.
-
Pregnenolone → Progesterone (SER): 3β-HSD oxidizes the hydroxyl group at C-3 to a ketone.
-
Progesterone → 17α-Hydroxyprogesterone (SER): 17α-hydroxylase (P-450c17/CYP17) adds -OH at C-17.
-
17α-Hydroxyprogesterone → 11-Deoxycortisol (SER): 21α-hydroxylase (P-450c21/CYP21A2) adds -OH at C-21.
-
11-Deoxycortisol → Cortisol (mitochondria): 11β-hydroxylase (P-450c11/CYP11B1) adds -OH at C-11.
Key enzyme absent from zona glomerulosa: 17α-hydroxylase is not substantially present in the glomerulosa, which is why that zone cannot make cortisol and instead makes aldosterone (using aldosterone synthase/CYP11B2). (Medical Physiology)
Clinical relevance - 21α-hydroxylase deficiency (most common congenital adrenal enzyme defect): Blocks synthesis of both cortisol and aldosterone. The resulting low cortisol removes negative feedback → ACTH rises → drives excessive production of adrenal androgens. Clinically: salt-wasting, hypoglycemia, and virilization (ambiguous genitalia in female infants).
3. Transport and Metabolism
-
~90% of circulating cortisol is protein-bound:
- ~75% bound to corticosteroid-binding globulin (CBG / transcortin) - high affinity, low capacity
- ~15% loosely bound to albumin
- Only ~10% is free (biologically active)
-
Synthetic glucocorticoids (dexamethasone, betamethasone, triamcinolone) have <1% affinity for CBG - they circulate largely free, contributing to greater biological activity per dose.
-
Cortisol is metabolized in the liver. Prednisone (prodrug) requires hepatic conversion to active prednisolone by 11β-HSD. In severe liver disease, prednisolone is preferred over prednisone. (Rheumatology, 2-Volume Set)
-
The enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) inactivates cortisol to cortisone in tissues like the kidney, protecting mineralocorticoid receptors from activation by cortisol (which has equal affinity for the mineralocorticoid receptor). Liquorice inhibits 11β-HSD2, causing apparent mineralocorticoid excess.
4. The HPA Axis: Regulation of Cortisol Secretion
The hypothalamic-pituitary-adrenal (HPA) axis controls cortisol secretion through a classic endocrine hierarchy:
Hypothalamus (paraventricular nucleus)
↓ CRH (41-amino acid neuropeptide)
Anterior Pituitary (corticotroph cells)
↓ ACTH (39-amino acid peptide)
Adrenal Cortex (zona fasciculata)
↓ Cortisol
⟲ Negative feedback → inhibits CRH & ACTH
CRH (Corticotropin-Releasing Hormone)
- Secreted by small-bodied neurons of the paraventricular nucleus (PVN) of the hypothalamus
- Released into the hypophyseal portal venous plexus → reaches anterior pituitary
- Binds CRH-R1 (GPCR) on corticotrophs → Gαs → ↑cAMP → PKA → opens L-type Ca²⁺ channels → exocytosis of pre-formed ACTH
- Over longer time: increases POMC gene transcription
- Arginine vasopressin (AVP) is a co-secretagogue from the PVN that potentiates CRH-induced ACTH release (important during stress involving dehydration or trauma)
ACTH (Adrenocorticotropic Hormone)
- A 39-amino acid peptide secreted by anterior pituitary corticotrophs
- Derived from pro-opiomelanocortin (POMC), a large precursor that also yields:
- β-lipotropin (β-LPH)
- α-MSH, γ-MSH (melanocyte-stimulating hormones - cause skin hyperpigmentation in Addison's disease via excess ACTH/POMC)
- CLIP (corticotropin-like intermediate lobe peptide)
- β-endorphin
- ACTH acts on adrenal fasciculata cells to stimulate cholesterol uptake and activate the rate-limiting step (P-450scc), rapidly driving cortisol synthesis
- Chronic ACTH excess causes adrenal hyperplasia; chronic ACTH deficiency causes adrenal atrophy
Negative Feedback
Cortisol exerts negative feedback at two levels:
- Anterior pituitary: Cortisol binds cytosolic GR → nucleus → binds GREs → inhibits POMC transcription and blocks synthesis of the CRH receptor; also inhibits release of pre-formed ACTH from vesicles.
- Hypothalamus: Cortisol decreases CRH mRNA levels in PVN neurons and inhibits release of pre-formed CRH (less dominant than pituitary feedback).
(Medical Physiology, Boron & Boulpaep)
5. Circadian Rhythm and Pulsatility
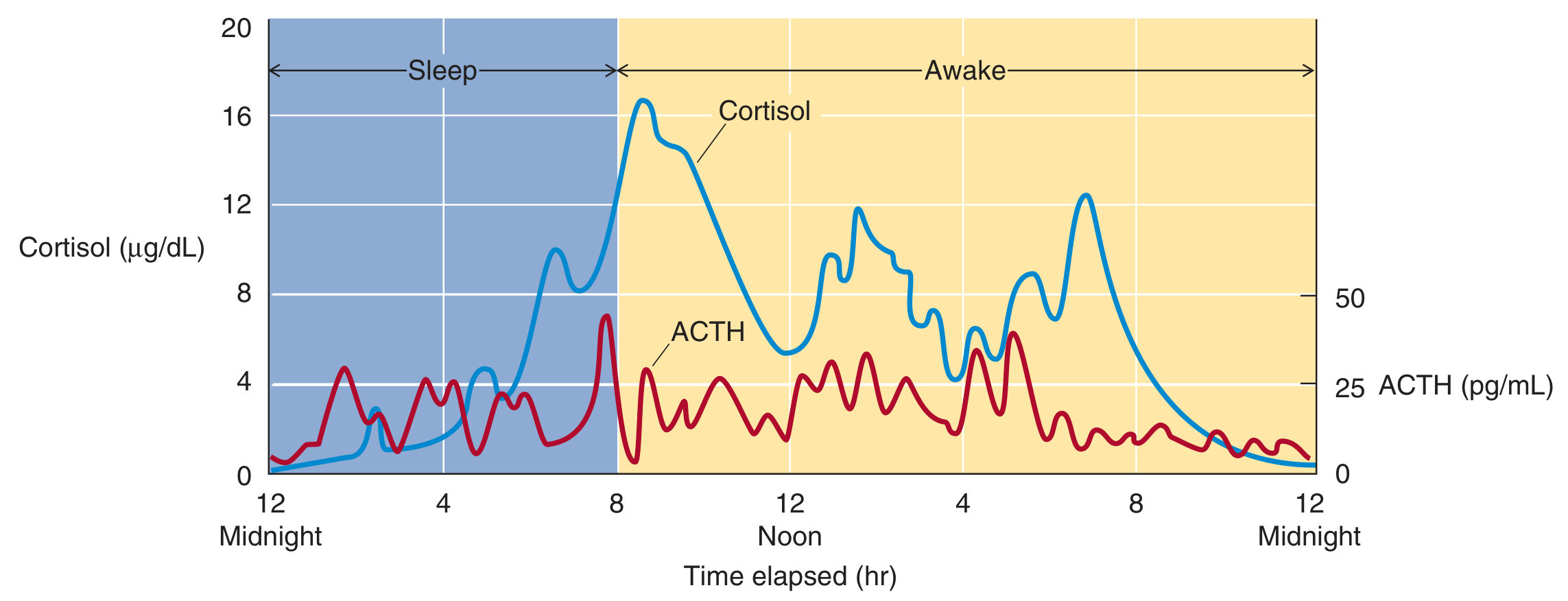
Figure: 24-hour rhythm of ACTH (red, pg/mL) and cortisol (blue, μg/dL). ACTH peaks in early morning around 8 AM with pulsatile bursts. Cortisol follows with its longer half-life, damping the pulsatile oscillations. Both approach nadir around midnight. (Medical Physiology)
Key features:
- ACTH secretory activity is greatest in the early morning and lowest late afternoon/evening
- Cortisol peaks at ~16 μg/dL around 8 AM and falls to near zero by midnight
- The rhythm is driven by the suprachiasmatic nucleus (receives retinal light input) - blind individuals lose circadian rhythms
- Superimposed on the circadian rhythm is pulsatile secretion driven by pulsatile CRH release
- ACTH pulses are sharper and briefer (shorter half-life); cortisol oscillations are broader and damped
6. Stress Response
Higher CNS centers stimulate the HPA axis during physical, psychological, and biochemical stress:
- Hypoglycemia → stimulates CRH and ACTH → ↑cortisol → gluconeogenesis raises blood glucose
- Physical trauma, infection, surgery, major illness → activate ascending noradrenergic pathways → stimulate PVN CRH neurons
- The amplitude of CRH secretory bursts increases during stress (not frequency)
- Chronic stress → chronic ACTH elevation → adrenal hypertrophy
- Glucocorticoids are sometimes called "stress hormones" because they allow the body to cope with acute threats - but chronic activation is harmful
7. Physiological Actions of Cortisol
A. Carbohydrate Metabolism - "Diabetogenic"
Cortisol is overall catabolic and diabetogenic:
- ↑ Gluconeogenesis in liver (key survival function during fasting)
- ↑ Protein catabolism in muscle → provides amino acids to liver as gluconeogenic substrates
- ↑ Lipolysis → provides glycerol to liver for gluconeogenesis; free fatty acids used as alternative fuel
- ↓ Glucose utilization by peripheral tissues (antagonizes insulin action)
- ↓ Insulin sensitivity of adipose tissue
In hypocortisolism (Addison's disease): hypoglycemia In hypercortisolism (Cushing's syndrome): hyperglycemia (Costanzo Physiology)
B. Protein Metabolism
- Catabolic in muscle, bone, connective tissue, skin - mobilizes amino acids
- Anabolic in the liver - stimulates synthesis of enzymes for gluconeogenesis
- Net effect of prolonged excess: muscle wasting, skin thinning, striae, poor wound healing
C. Fat Metabolism - Redistribution
- Excess cortisol causes characteristic fat redistribution:
- ↑ Fat deposition: face (moon facies), trunk, supraclavicular, dorsal interscapular (buffalo hump)
- ↓ Fat in extremities
- Mechanism: differential insulin sensitivity across fat depots; visceral fat is more glucocorticoid-sensitive
D. Anti-Inflammatory Actions
Three major mechanisms:
- Induces lipocortin (annexin A1) - inhibits phospholipase A2, blocking release of arachidonic acid from membrane phospholipids → suppresses prostaglandin and leukotriene synthesis
- Inhibits IL-2 production and T lymphocyte proliferation
- Inhibits histamine and serotonin release from mast cells and platelets
(Costanzo Physiology)
E. Immune Suppression
- Suppresses cellular immunity via T-cell inhibition (IL-2 ↓)
- Reduces Fc receptor expression on macrophages
- Promotes neutrophilia (demargination from vessel walls) while impairing neutrophil migration to inflammatory sites
- Basis for glucocorticoid use in transplant rejection and autoimmune diseases
F. Cardiovascular - Permissive Role
Cortisol is required for normal vascular tone and blood pressure maintenance:
- Up-regulates α₁-adrenergic receptors on arterioles (permissive effect)
- Allows arterioles to respond normally to catecholamines (norepinephrine, epinephrine)
- Without cortisol → hypotension (hallmark of Addison's disease - refractory to vasopressors)
- Excess cortisol → hypertension (Cushing's syndrome)
(Costanzo Physiology)
G. Bone and Calcium Metabolism
Cortisol has anti-anabolic/catabolic effects on bone:
- Decreases synthesis of type I collagen (major bone matrix component)
- Decreases osteoblast function (bone formation ↓)
- Decreases intestinal Ca²⁺ absorption (interferes with vitamin D action)
- Net result: negative calcium balance and osteoporosis with chronic excess
H. Renal Effects
- Increases GFR by causing afferent arteriole vasodilation → ↑ renal blood flow
- Without cortisol, GFR falls; excess cortisol can contribute to hypertension via this mechanism and via sodium retention (weak mineralocorticoid effect of cortisol)
I. CNS Effects
- Glucocorticoid receptors are widely distributed in the CNS (hippocampus, prefrontal cortex, hypothalamus)
- Normal cortisol: needed for normal mood, cognition, and arousal
- Excess cortisol: euphoria, emotional lability, depression, psychosis, insomnia, decreased REM sleep
- Deficiency: fatigue, depression, cognitive impairment
8. Summary Table: Physiological Actions of Glucocorticoids
| System | Effect |
|---|---|
| Carbohydrate | ↑ Gluconeogenesis, ↓ peripheral glucose uptake, ↑ glycogen storage (liver) |
| Protein | ↑ Catabolism (muscle), ↑ hepatic protein synthesis for gluconeogenesis |
| Fat | ↑ Lipolysis; redistribution of fat (central deposition) |
| Immune | ↓ IL-2, ↓ T-cell proliferation, ↓ histamine release, ↓ prostaglandins |
| Cardiovascular | Permissive for catecholamine vasoconstriction (↑ α₁-adrenergic receptors) |
| Bone | ↓ Osteoblast activity, ↓ collagen synthesis, ↓ intestinal Ca²⁺ absorption → osteoporosis |
| Kidney | ↑ GFR (afferent arteriole vasodilation) |
| CNS | Mood, cognition, arousal; excess → emotional disturbance, ↓ REM sleep |
| HPA axis | Negative feedback on CRH and ACTH secretion |
(Costanzo Physiology 7th ed.; Medical Physiology, Boron & Boulpaep)
9. Clinical Correlates
| Condition | Mechanism | Key Features |
|---|---|---|
| Addison's disease (primary adrenal insufficiency) | Adrenal destruction (autoimmune most common today; formerly TB) | Fatigue, hypoglycemia, hypotension, hyponatremia, hyperkalemia, hyperpigmentation (↑ACTH/MSH), salt craving |
| Cushing's syndrome | Glucocorticoid excess (exogenous steroids, pituitary ACTH tumor, adrenal tumor, ectopic ACTH) | Central obesity, moon facies, buffalo hump, hypertension, hyperglycemia, osteoporosis, striae, easy bruising |
| Cushing's disease | Pituitary ACTH-secreting adenoma specifically | Bilateral adrenal hyperplasia, non-suppressible on low-dose dexamethasone test |
| Congenital adrenal hyperplasia | 21α-hydroxylase deficiency (most common) | Salt-wasting, hypoglycemia, virilization (↑adrenal androgens) |
| Exogenous steroid withdrawal | HPA axis suppression → adrenal atrophy | Adrenal crisis on abrupt cessation; must taper slowly |
This is a shared conversation. Sign in to Orris to start your own chat.