Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive, detailed mechanistic content from Murray & Nadel's Textbook of Respiratory Medicine. I have everything needed to write a thorough explanation. Let me compose the full answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is a syndrome of noncardiogenic pulmonary edema driven by widespread inflammatory lung injury. Unlike cardiogenic pulmonary edema, the alveolar fluid in ARDS is exudative - protein-rich and not simply the result of elevated hydrostatic pressure. The underlying pathology is diffuse alveolar damage (DAD), and the mechanism unfolds through several interconnected pathways.
1. The Berlin Definition (Diagnostic Framework)
The 2012 Berlin definition requires:
-
Bilateral opacities on chest X-ray or CT not fully explained by effusions, collapse, or nodules
-
PaO2/FiO2 ratio <300 mmHg (mild: <300; moderate: <200; severe: <100)
-
Onset within 7 days of a known clinical insult
-
Respiratory failure not fully explained by cardiac failure or fluid overload
-
Sabiston Textbook of Surgery, p. 940
2. Inciting Causes
ARDS can be triggered by direct (pulmonary) or indirect (extrapulmonary) insults:
| Direct | Indirect |
|---|---|
| Pneumonia | Sepsis |
| Aspiration | Severe trauma/hemorrhage |
| Pulmonary contusion | Pancreatitis |
| Inhalation injury | Blood transfusions (TRALI) |
| Near-drowning | Cardiopulmonary bypass |
- Murray & Nadel's Textbook of Respiratory Medicine
3. The Three Pathological Phases
Phase 1 - Exudative Phase (Days 1-7)
- Hyaline membranes form from cellular debris, proteins, and denatured surfactant components
- Protein-rich fluid fills alveolar spaces due to barrier breakdown
- Widespread epithelial and endothelial disruption
- Massive neutrophil infiltration of the interstitium and airspaces
- This is termed Diffuse Alveolar Damage (DAD)
Phase 2 - Proliferative Phase (Days 7-21)
- Hyaline membranes are reorganized
- Early fibrosis appears
- Obliteration of pulmonary capillaries
- Deposition of interstitial and alveolar collagen
- Neutrophil numbers decrease
Phase 3 - Fibrotic Phase (>2 weeks)
-
Pulmonary fibrosis develops in a subset of patients
-
N-terminal procollagen peptide III (a collagen synthesis marker) can be detected in BAL fluid as early as 24 hours, suggesting fibroproliferation begins simultaneously with inflammatory injury, not after it
-
Murray & Nadel's Textbook of Respiratory Medicine, p. 3074-3076
Important caveat: Only ~50% of ARDS patients show DAD on autopsy or biopsy. Patients with confirmed DAD tend to have worse compliance, lower PaO2/FiO2, and roughly 5x greater risk of death from hypoxemic failure.
4. The Alveolar-Capillary Barrier
The alveolar-capillary barrier has two cellular layers:
Endothelial cells - line the pulmonary capillaries; disruption increases vascular permeability
Epithelial cells - the alveolar lining:
- Type I pneumocytes cover ~95% of the alveolar surface; extremely vulnerable to injury
- Type II pneumocytes cover ~5% of the surface; produce surfactant and serve as progenitors for type I cells during repair
In ARDS, both layers are injured. The epithelial barrier is actually more critical to the outcome than the endothelial barrier, because:
- A more permeable epithelium allows flooding of the normally dry airspace
- Type II cell dysfunction impairs surfactant production and active ion transport (Na⁺/K⁺-ATPase, ENaC), which is the primary mechanism for alveolar fluid clearance
- The degree of type II cell impairment correlates with severity and outcome
- Murray & Nadel's Textbook of Respiratory Medicine, p. 3083+
5. Neutrophil-Mediated Injury
Neutrophils are the primary effectors of alveolar damage:
- Recruitment: Inflammatory cytokines (IL-8/CXCL8, TNF-α, IL-1β, IL-6) and complement fragments (C5a) massively recruit neutrophils into the pulmonary capillaries, interstitium, and airspaces
- Activation: Activated neutrophils release a destructive arsenal:
- Neutrophil elastase (NE) - cleaves extracellular matrix proteins, tight junction proteins, and surfactant proteins; degrades structural integrity of the barrier
- Matrix metalloproteinases (MMPs) - further degrade the basement membrane
- Reactive oxygen species (ROS) - oxidative damage to lipid membranes, proteins, and DNA
- Myeloperoxidase (MPO) - generates hypochlorous acid (bleach-like oxidant)
- Platelet-activating factor (PAF) - amplifies inflammation and increases vascular permeability
- NETosis: Neutrophils form neutrophil extracellular traps (NETs) which contribute to microvascular occlusion and barrier injury
PI3-kinase-γ signaling is a key intracellular pathway governing neutrophil accumulation - knockout mice show markedly decreased neutrophil recruitment and lung injury.
- Murray & Nadel's Textbook of Respiratory Medicine, p. 3140-3151
6. Surfactant Dysfunction
-
Surfactant normally reduces alveolar surface tension, prevents collapse, and has anti-inflammatory properties
-
In ARDS: plasma proteins (fibrinogen, albumin, immunoglobulins) flooding the alveolus competitively inhibit surfactant function
-
Phospholipase A2 (released during pancreatitis, among other conditions) enzymatically degrades surfactant
-
Type II pneumocyte injury directly reduces surfactant synthesis and secretion
-
Result: alveolar collapse at end-expiration, increased work of breathing, worsening hypoxemia, decreased lung compliance
-
Murray & Nadel's Textbook of Respiratory Medicine
7. Coagulation and Fibrin Deposition
The inflammatory and coagulation systems are deeply intertwined in ARDS:
- Alveolar and intravascular coagulation is activated
- Fibrin deposition occurs in alveolar spaces (contributing to hyaline membrane formation) and in pulmonary microvessels
- Plasminogen activator inhibitor-1 (PAI-1) is markedly elevated, suppressing fibrinolysis and promoting fibrin persistence
- Elevated PAI-1 is a biomarker of the hyperinflammatory ARDS subphenotype and correlates with worse mortality
- Pulmonary microvascular occlusion from fibrin thrombi increases dead space ventilation and pulmonary hypertension
8. Angiopoietin Pathway
The angiopoietin-Tie2 axis regulates vascular stability:
- Angiopoietin-1 (Ang-1): Produced by pericytes; binds Tie2 and maintains endothelial quiescence and barrier integrity
- Angiopoietin-2 (Ang-2): Stored in Weibel-Palade bodies; released rapidly by endothelial activation; acts as an Ang-1 antagonist; destabilizes the endothelium and promotes vascular permeability and neutrophil adhesion
- In ARDS: plasma Ang-2 is markedly elevated and correlates with severity and mortality
- Ang-2 also acts synergistically with TNF-α to upregulate endothelial adhesion molecules, amplifying neutrophil trafficking
9. Na⁺/Water Clearance Failure
Under normal conditions, the alveolar epithelium actively clears fluid by:
- ENaC (epithelial sodium channel) absorbing Na⁺ from the lumen
- Na⁺/K⁺-ATPase on the basolateral surface pumping Na⁺ out
- Water follows osmotically through aquaporins
In ARDS, multiple factors impair this clearance:
- Direct type II cell damage
- Hypoxia downregulates Na⁺ transporter expression
- Cytokines (TNF-α, TGF-β) inhibit ion transport
- Catecholamine-stimulated upregulation of transporters is blunted
The net result is persistent alveolar flooding despite resolution of initial injury triggers.
10. Physiological Consequences
| Abnormality | Mechanism |
|---|---|
| Severe hypoxemia (refractory to O2) | Intrapulmonary right-to-left shunt through fluid-filled, non-ventilated alveoli |
| Decreased compliance | Alveolar flooding, surfactant loss, atelectasis, fibrosis |
| Increased dead space | Microvascular occlusion, pulmonary vasoconstriction |
| Pulmonary hypertension | Hypoxic vasoconstriction, fibrin thrombi, vessel compression by PEEP |
| Increased work of breathing | Stiff lungs, increased respiratory drive |
The classic chest X-ray/CT appearance is bilateral, diffuse, patchy opacities with a gravitational gradient - dependent zones are consolidated/fluid-filled, while non-dependent zones are relatively spared (the "sponge" model).
11. Ventilator-Induced Lung Injury (VILI)
Mechanical ventilation, while life-saving, can worsen ARDS through:
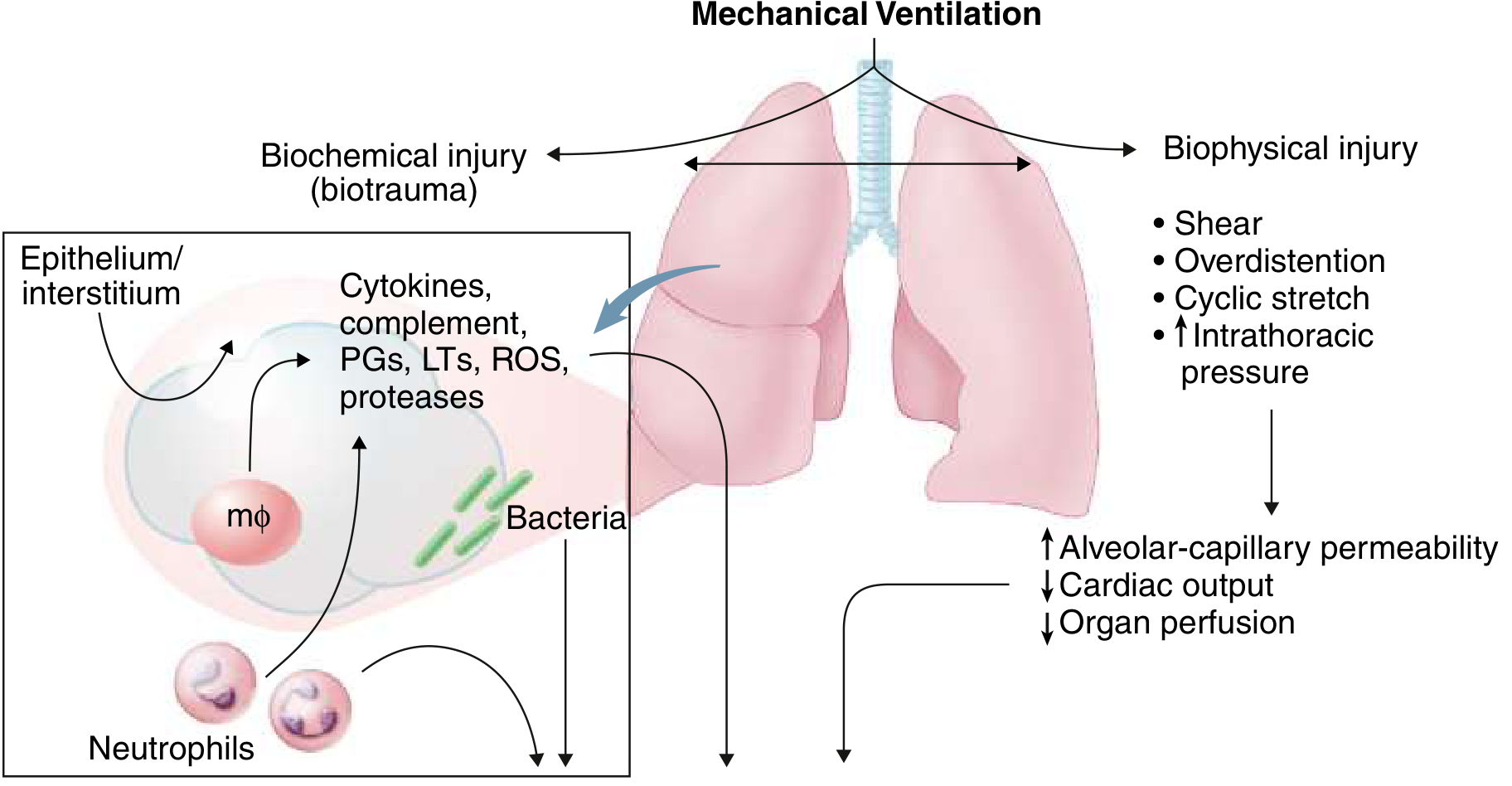
- Volutrauma: Overdistention of non-collapsed alveoli (the lung in ARDS is "baby-sized" - only ~30% of normal alveoli remain open)
- Atelectrauma: Cyclic opening and closing of collapsed units generates shear stress
- Barotrauma: Excessive pressures rupture alveoli
- Biotrauma: Mechanical stretch induces cytokine release (mechano-transduction), amplifying systemic inflammation and contributing to multi-organ failure
This underpins the ARDSNet lung-protective ventilation strategy: low tidal volume (6 mL/kg predicted body weight), plateau pressure ≤30 cmH2O, and appropriate PEEP.
- Goldman-Cecil Medicine, p. 1066-1067
12. ARDS Subphenotypes
Latent class analysis of large ARDSNet trial datasets reveals two biological subphenotypes:
| Feature | Subphenotype 1 (Hypoinflammatory) | Subphenotype 2 (Hyperinflammatory) |
|---|---|---|
| IL-6, IL-8, TNF-sR1 | Low | High |
| PAI-1 | Low | High |
| Protein C | Higher | Lower |
| Vasopressors | Less common | More common |
| Predominant cause | Trauma | Sepsis |
| Mortality | Lower | Higher (~50%) |
| Best PEEP strategy | Low | High |
These subphenotypes respond differently to fluid management and PEEP strategies, highlighting the pathophysiological heterogeneity underlying the ARDS label.
- Murray & Nadel's Textbook of Respiratory Medicine, p. 3271-3272
Summary Diagram
Triggering insult (sepsis, pneumonia, trauma, etc.)
↓
Systemic/local inflammatory cascade
(TNF-α, IL-1β, IL-6, IL-8, C5a, PAF)
↓
Neutrophil sequestration in lung
↓
Release of elastase, ROS, MMPs, MPO
↓
Endothelial + Epithelial barrier disruption
↓
┌──────────────────────────────────────┐
│ Protein-rich exudate floods alveoli │
│ Surfactant inactivation │
│ Type II pneumocyte dysfunction │
│ Fibrin deposition (hyaline membranes)│
│ Impaired Na⁺/fluid clearance │
└──────────────────────────────────────┘
↓
DIFFUSE ALVEOLAR DAMAGE
↓ Compliance | ↑ Shunt | Hypoxemia
Pulmonary hypertension | ↑ Dead space
↓
ARDS (Berlin criteria met)
± VILI from mechanical ventilation
± Fibroproliferative phase → Fibrosis
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine (2-Volume), pp. 3064-3275
- Goldman-Cecil Medicine International Edition, pp. 1065-1068
- Sabiston Textbook of Surgery (Biological Basis of Modern Surgical Practice), p. 940
This is a shared conversation. Sign in to Orris to start your own chat.