Give me details answer on approach to inborn error of metabolism from stardard paediatric textbook
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive content from authoritative textbooks with diagnostic flowcharts. Let me compile the complete, detailed answer.
Approach to Inborn Errors of Metabolism (IEM) — Paediatric Perspective
Sources: Goldman-Cecil Medicine (Chapter 189), Tintinalli's Emergency Medicine (Chapter 146), Bradley and Daroff's Neurology in Clinical Practice (Chapter 110)
1. Definition and Classification
Inborn errors of metabolism (IEM) include not only enzyme deficiencies but any condition where perturbation of a biochemical pathway is intrinsic to the pathobiology of the disorder. The >1,400 known IEM can be classified into three broad groups:
| Category | Mechanism | Examples |
|---|---|---|
| Small molecule / intoxication disorders | Accumulation of toxic metabolites upstream of the enzyme block | Aminoacidopathies, organic acidurias, urea cycle defects |
| Complex molecule disorders | Abnormal accumulation of glycogen, sphingolipids, glycosaminoglycans | Lysosomal storage diseases, MPS, peroxisomal disorders |
| Energy deficiency disorders | Impaired energy production | Mitochondrial respiratory chain defects, fatty acid oxidation disorders |
Inheritance patterns include autosomal recessive (most common), autosomal dominant, X-linked (e.g., OTC deficiency), and mitochondrial (matrilineal).
Epidemiology: Each disorder is individually rare but collectively the incidence is as high as 1 per 800 live births, making IEM as a group relatively common.
2. Pathophysiology
Most IEM result from single-gene defects causing:
- Accumulation of toxic substrates upstream of the impaired enzyme
- Accumulation of toxic byproducts via alternate metabolic pathways
- Deficiency of the reaction product (downstream)
- Vitamin cofactor deficiencies (e.g., B12 required for methylmalonyl-CoA mutase)
Because most metabolic toxins cross the placenta and are cleared by maternal enzymes, most newborns are asymptomatic at birth and present after a variable delay once enteral feeding begins.
Key exception: Nonketotic hyperglycinemia — presents at birth with encephalopathy, hypotonia, myoclonic seizures, and corpus callosum dysplasia, because glycine acts in utero.
Disorders of intermediary metabolism (aminoacidopathies) produce acute or chronic intoxication, while complex molecule disorders (MPS, peroxisomal) have a more chronic and progressive course.
3. Clinical Presentation
Neonatal / Infant Presentation ("Small Molecule" Disorders)
The acute presentation is often nonspecific and mimics sepsis:
- Irritability, lethargy, vomiting, poor feeding
- Seizures, apnoea, altered consciousness, coma
- Metabolic acidosis, hypoglycaemia
- Hypothermia (especially in urea cycle defects and organic acidaemias)
- Tachypnoea without increased work of breathing (Kussmaul breathing from metabolic acidosis → respiratory alkalosis in urea cycle defects)
Important historical clues:
- Symptoms worse in the morning before first feed (fasting intolerance)
- Parental aversion to protein or carbohydrates
- Recurrent hospitalisations responding to IV fluids and glucose
- Previous unexplained neonatal deaths, spontaneous abortions in the family
- Maternal acute fatty liver of pregnancy or HELLP syndrome → suggests heterozygosity for fatty acid oxidation defects
- Unusual body or urinary odour:
- Sweaty feet → isovaleric acidaemia, glutaric acidaemia
- Maple syrup → maple syrup urine disease (MSUD)
- Musty → phenylketonuria
Later Childhood Presentation (Complex Molecule / Storage Disorders)
Present with progressive, insidious features:
- Developmental delay, regression, neurodegeneration
- Dysmorphic / coarse facial features
- Hepatosplenomegaly
- Skeletal dysplasia
4. Organ-Specific Clinical Signs
| Organ | Clinical Sign | Disorder |
|---|---|---|
| Eye (cornea) | Corneal clouding/dystrophy | MPS (Hurler, Maroteaux-Lamy, Sly) |
| Eye (lens) | Dislocated lens | Homocystinuria |
| Eye | Cataracts | Galactosaemia |
| Skeletal | Ochronosis, black urine | Alkaptonuria |
| CNS | Ataxia, ID, seizures, peripheral neuropathy | Respiratory chain deficiency, CDG |
| Heart | Cardiomyopathy | Fabry disease, LCFAO disorders, GSD |
| Muscle | Hypotonia, rhabdomyolysis | Pompe disease, fatty acid oxidation |
| Liver | Hepatomegaly/splenomegaly, cirrhosis | MPS, GSD, Gaucher, Niemann-Pick |
| Kidney | Renal insufficiency, proteinuria | Cystinosis, Fabry, MMA |
| Skin | Angiokeratoma, hypopigmentation | Fabry disease, PKU |
| GU | Female virilisation | Congenital adrenal hyperplasia |
5. Diagnostic Approach
The key principle is: making a definitive diagnosis is not as important as maintaining a high index of suspicion. Acute stabilisation can precede definitive diagnosis.
First-Tier (Bedside / Emergency) Investigations
| Test | Interpretation |
|---|---|
| Blood glucose | Hypoglycaemia → GSD, FAOD, organic acidaemias, gluconeogenesis defects |
| Urine/serum ketones | Hypoglycaemia + no ketones → fatty acid oxidation defect; hypoglycaemia + elevated ketones → organic acidaemia |
| Plasma ammonia | Normal neonates: <65 µmol/L; >200 µmol/L = metabolic disease; >400 µmol/L = urea cycle defect |
| Blood gas | Metabolic acidosis ± anion gap |
| Anion gap (Na − [Cl + HCO₃]) | >15 → organic acidaemias (often >30–50 mEq/L with ketosis) |
| Plasma lactate | Elevated in mitochondrial disorders, pyruvate metabolism defects, GSD |
| Serum electrolytes | Electrolyte disturbances |
Second-Tier Investigations
| Test | Diagnostic Implication |
|---|---|
| CBC | Pancytopenia → propionic/isovaleric/methylmalonic acidaemia |
| Liver function tests | Unconjugated bilirubin → galactosaemia; hepatic failure → FAOD, mitochondrial, urea cycle |
| Creatine kinase | Elevated → mitochondrial disorders |
| Serum amino acids | Aminoacidopathies, urea cycle defects, mitochondrial disorders |
| Serum acylcarnitine profile | Organic acidurias, FAOD, mitochondrial disorders, carnitine deficiency |
| Urine organic acids | Organic acidaemias, FAOD, mitochondrial, peroxisomal |
| Urine reducing substances | Positive → galactosaemia, tyrosinaemia |
| Urine orotic acid | Elevated → OTC deficiency (urea cycle) |
| Urine acylglycines | Aminoacidopathies, organic acidurias |
| CSF lactate/pyruvate/amino acids | Mitochondrial disease, nonketotic hyperglycinaemia |
Third-Tier / Definitive Investigations
- Plasma amino acids + acylcarnitine profile + urine organic acids (the trio for metabolic crisis work-up)
- Metabolomics — untargeted analysis of hundreds of metabolites from single blood/urine/CSF sample; improves diagnostic rates over traditional testing
- DNA molecular analysis / exome or genome sequencing — now largely replaced tissue biopsies; provides genotype-phenotype information and informs counselling
6. Diagnostic Algorithm
The two key initial questions are:
- Is there acidosis?
- Is there elevated ammonia?
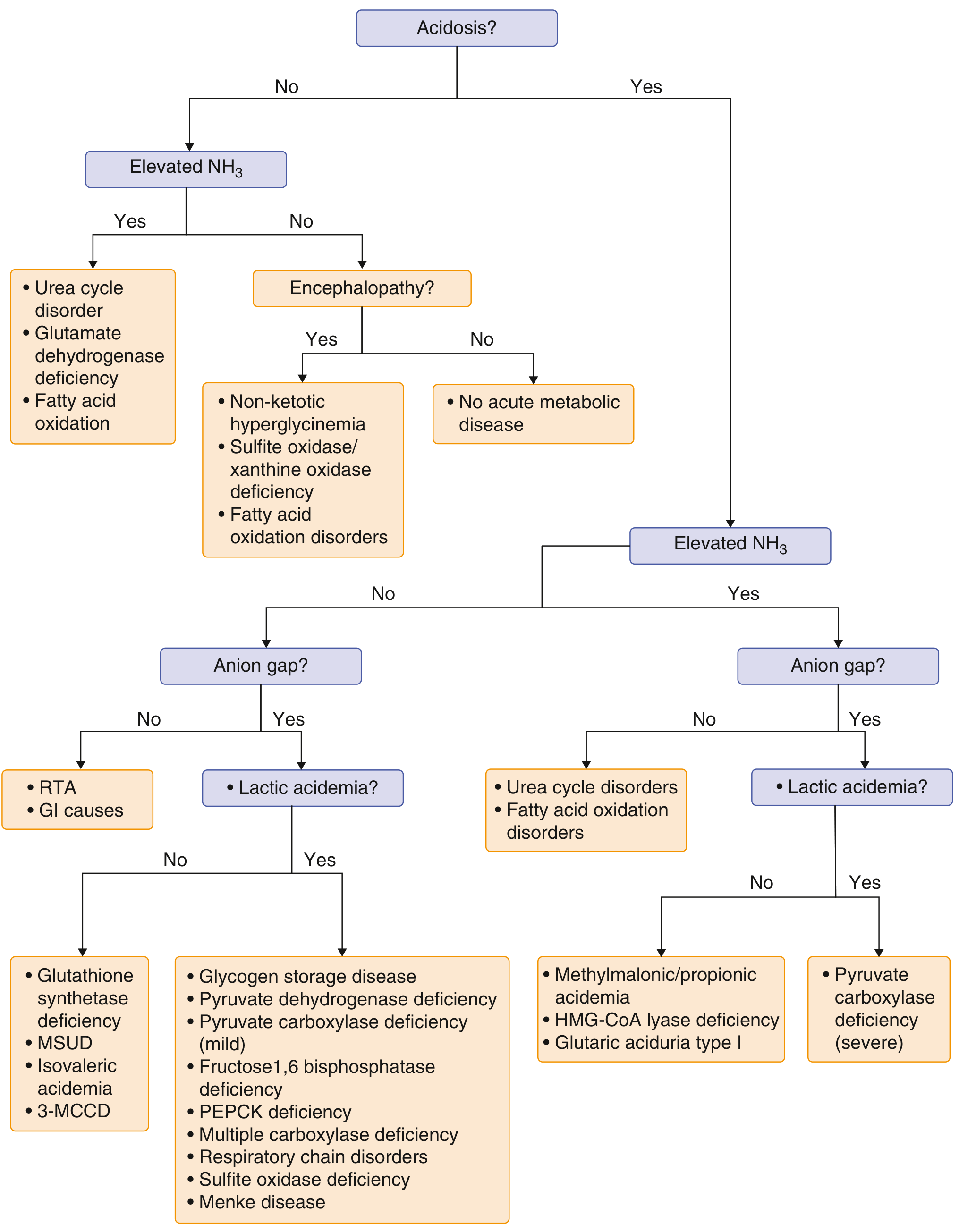
Simplified bedside algorithm:
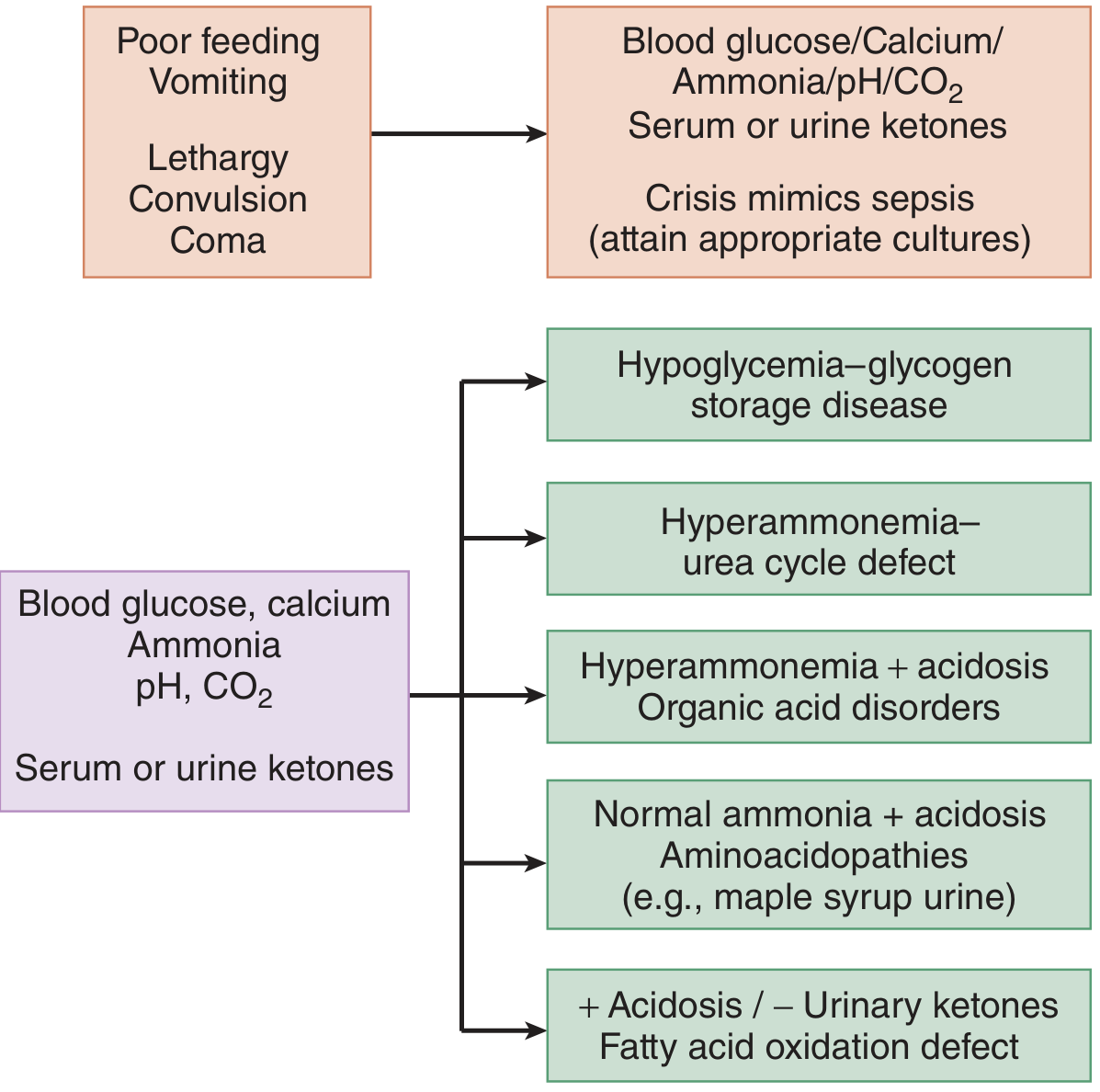
| Pattern | Think of |
|---|---|
| Hypoglycaemia, no ketones | Fatty acid oxidation defect |
| Hypoglycaemia + ketones | Organic acidaemia, GSD |
| Hyperammonaemia, NO acidosis | Urea cycle defect |
| Hyperammonaemia + acidosis | Organic acidaemia |
| Elevated anion gap acidosis + lactic acidosis | Mitochondrial / pyruvate metabolism disorders |
| Elevated anion gap acidosis, NO lactic acidosis | MSUD, isovaleric acidaemia, organic acidaemias |
| Normal ammonia + acidosis | Aminoacidopathies (e.g., MSUD) |
| Normal ammonia, anion gap, NO lactic acidosis | RTA, GI causes |
7. Treatment — Acute Management
Despite the diversity of IEM, emergency resuscitation is relatively straightforward and follows common principles.
Goals
- Remove the inciting metabolic substrate (stop breast milk/formula)
- Provide energy substrate to halt catabolism and toxin production
- Restore circulatory volume and electrolyte balance
- Clear toxic metabolites (e.g., organic acids, ammonia)
ABCDs First
- Airway, breathing, circulation, disability (neurological status)
- Apnoea/hypoventilation → positive-pressure ventilation, intubation, supplemental oxygen
- Respiratory acidosis worsens metabolic acidosis if ventilation is inadequate
Fluid Resuscitation
- Crystalloid bolus: 10–20 mL/kg in neonates, 20 mL/kg in infants — reassess frequently
- Even non-shocked patients benefit from normal saline bolus + double maintenance with dextrose
- Dextrose provides metabolic substrate; aggressive hydration promotes urinary clearance of toxic metabolites
- Avoid hypotonic fluids → risk of cerebral oedema, especially in hyperammonaemic states
Dextrose (for Hypoglycaemia)
| Age | Bolus | Maintenance |
|---|---|---|
| Neonate | D10, 5 mL/kg IV/IO | 6 mL/kg/h D10 |
| Infant | D10, 5 mL/kg or D25 2 mL/kg | 6 mL/kg/h D10 |
| Child | D25, 2 mL/kg | 6 mL/kg/h D10 (adjust by weight) |
| Adolescent | D25 or D50 1 mL/kg | As for child |
Glucagon 0.03 mg/kg IM and hydrocortisone 25–50 mg can also be considered as adjuncts.
Hyperammonaemia Management
- Stop all protein intake
- High-calorie glucose/lipid infusion to suppress catabolism
- For urea cycle defects: sodium benzoate + sodium phenylacetate (nitrogen scavengers)
- Dialysis for severe, refractory hyperammonaemia (>500 µmol/L) causing encephalopathy
Treat Concurrent Sepsis
- Because IEM metabolic crisis is clinically indistinguishable from sepsis, empirical broad-spectrum antibiotics are mandatory until sepsis is excluded:
- Neonates: ampicillin + gentamicin
- Infants 28–90 days: cefotaxime
- Older infants/children: ceftriaxone
Seizure Management
- Standard antiepileptics
- Pyridoxine (B6) empirically if seizures refractory → consider pyridoxine-dependent epilepsy or B6-responsive conditions
8. Newborn Screening
Tandem mass spectrometry (MS/MS)-based newborn screening has dramatically expanded the detection of IEM before symptom onset. Conditions now included in expanded newborn screening panels include:
- Phenylketonuria (PKU)
- MSUD
- Glutaric acidaemia type I
- Methylmalonic acidaemia, propionic acidaemia
- Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency
- Tyrosinaemia type I
- Galactosaemia
- Homocystinuria
9. Long-Term Management Principles
| Strategy | Examples |
|---|---|
| Dietary restriction | Low-phenylalanine diet (PKU); low-protein diet (urea cycle defects, organic acidaemias) |
| Metabolic formula | Amino acid-based formulae lacking the offending amino acid |
| Cofactor supplementation | B12 for methylmalonyl-CoA mutase; B6 for homocystinuria; biotin for multiple carboxylase deficiency |
| Substrate reduction | N-acetylglutamate for urea cycle |
| Enzyme replacement therapy | Gaucher disease (imiglucerase), Fabry disease (agalsidase), Pompe disease (alglucosidase) |
| Haematopoietic stem cell transplant | MPS I (Hurler), adrenoleukodystrophy |
| Liver transplantation | Tyrosinaemia type I, MSUD, propionic acidaemia |
| Gene therapy | Emerging; clinical trials underway for several IEM |
| Pharmacological chaperones | Migalastat for Fabry disease (amenable mutations) |
10. Selected Important IEM — Quick Reference
| Disorder | Defect | Key Features |
|---|---|---|
| PKU | Phenylalanine hydroxylase | ID, hypopigmentation, seizures, musty odour; treat with low-Phe diet ± sapropterin (BH4) |
| MSUD | Branched-chain α-keto acid dehydrogenase | Encephalopathy, ataxia, brain herniation; maple syrup odour |
| OTC deficiency | Ornithine transcarbamylase | X-linked; severe hyperammonaemia |
| Propionic acidaemia | Propionyl-CoA carboxylase | Neonatal metabolic acidosis, hyperammonaemia, dystonia, cardiomyopathy |
| Methylmalonic acidaemia | Methylmalonyl-CoA mutase | Acidosis, hyperammonaemia, renal failure, optic atrophy |
| Galactosaemia | Gal-1-P uridylyltransferase | Jaundice, hepatic failure, cataracts, hypoglycaemia, E. coli sepsis |
| Tyrosinaemia type I | Fumarylacetoacetate hydrolase | Acute/chronic liver failure, tubulopathy, hepatocellular carcinoma |
| Gaucher disease | Glucocerebrosidase | Hepatosplenomegaly, anaemia, thrombocytopenia, osteoporosis |
| Nonketotic hyperglycinaemia | Glycine cleavage enzyme | Neonatal encephalopathy, hypotonia, severe seizures, corpus callosum dysplasia |
| MCAD deficiency | Medium-chain acyl-CoA dehydrogenase | Hypoketotic hypoglycaemia on fasting; SIDS-like; treat with avoid fasting |
Key Teaching Points
- IEM is a diagnosis to actively consider, not a last resort — index of suspicion is the most important tool.
- The neonatal presentation of IEM is clinically indistinguishable from sepsis — treat both simultaneously.
- The initial 5 tests (glucose, ketones, ammonia, blood gas, lactate) direct acute management before confirmatory results are available.
- Stop feeds / protein in any sick neonate/infant suspected of IEM — this alone can prevent worsening intoxication.
- Dextrose is both diagnostic and therapeutic — it terminates catabolism and provides alternative energy.
- Newborn screening has transformed prognosis — many severe IEM are now diagnosed and treated pre-symptomatically.
References: Goldman-Cecil Medicine, 27th ed., Chapter 189 (Craigen WJ) | Tintinalli's Emergency Medicine: A Comprehensive Study Guide, 9th ed., Chapter 146 | Bradley and Daroff's Neurology in Clinical Practice, 8th ed., Chapter 110
This is a shared conversation. Sign in to Orris to start your own chat.