Cml
Reading File
Finding Sources
Searching PubMed
"chronic myeloid leukemia" AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have rich, comprehensive data from multiple authoritative sources. Let me compile the full CML overview.
Chronic Myeloid Leukemia (CML)
Definition
CML is a myeloproliferative neoplasm originating from a pluripotent hematopoietic stem cell. It is defined by a specific molecular lesion: the BCR-ABL fusion gene, which encodes a constitutively active 210 kDa tyrosine kinase. There is no BCR-ABL-negative CML - diseases without this fusion (e.g., "atypical CML," chronic neutrophilic leukemia) are distinct entities.
- Goldman-Cecil Medicine, p. 1938; Robbins Pathologic Basis of Disease, p. 583
Molecular Pathogenesis
| Feature | Detail |
|---|---|
| Cytogenetic event | Reciprocal translocation t(9;22)(q34;q11) |
| Result | Shortened chromosome 22 = Philadelphia (Ph) chromosome; elongated chromosome 9 |
| Fusion gene | BCR::ABL - constitutively active tyrosine kinase (210 kDa) |
| Frequency | >90% of cases show the Ph chromosome by karyotype; remainder detected by FISH or PCR |
| Cell of origin | Pluripotent HSC (hence both myeloid and lymphoid blast crisis are possible) |
Mechanism: The BCR dimerization domain causes constitutive self-association, activating the ABL kinase moiety. This drives RAS and JAK/STAT signaling - the same pathways activated by normal hematopoietic growth factors - causing granulocytic and megakaryocytic proliferation and premature release of immature cells from the marrow.
- Robbins Pathologic Basis of Disease, p. 583
Epidemiology
- Peak incidence: 5th-6th decade of life
- ~4,800 new cases/year in the United States
- Occurs in children and adolescents, but primarily an adult disease
- Without treatment: median survival ~3 years
- With TKI therapy: near-normal age-adjusted lifespan in chronic-phase disease
Disease Phases
| Phase | ELN Criteria | WHO Criteria |
|---|---|---|
| Chronic | Blasts <15% (peripheral/marrow) | Blasts <10% |
| Accelerated | Blasts 15-<30% peripheral/marrow | Blasts 10-<20% |
| Blast crisis | Blasts ≥30% | Blasts ≥20% |
75% of patients in developed countries are diagnosed in chronic phase.
Natural progression (untreated):
-
After ~3 years, ~50% enter accelerated phase (rising anemia, thrombocytopenia, basophilia, new cytogenetic abnormalities - trisomy 8, isochromosome 17q, Ph duplication)
-
Accelerated phase terminates in blast crisis within 6-12 months
-
Other 50% progress directly to blast crisis without an accelerated phase
-
Blast crisis types: 70% myeloid, ~30% lymphoid (pre-B cell) - confirming pluripotent stem cell origin
-
Goldman-Cecil Medicine, p. 1940; Robbins Pathologic Basis of Disease, p. 583
Morphology
Bone marrow:
- Markedly hypercellular with massively increased maturing granulocytic precursors
- Elevated proportion of eosinophils and basophils
- Increased megakaryocytes (often small, dysplastic)
- Characteristic sea-blue histiocytes (scattered macrophages with wrinkled green-blue cytoplasm)
- Increased reticulin; overt fibrosis is rare
Peripheral blood:
- Leukocytosis often >100,000 cells/µL
- Spectrum: neutrophils, band forms, metamyelocytes, myelocytes, eosinophils, basophils
- Blasts usually <10% of circulating cells
- Thrombocytosis (often marked)
Other:
-
Massive splenomegaly (extramedullary hematopoiesis, often with splenic infarcts)
-
Mild hepatomegaly and lymphadenopathy
-
Robbins Pathologic Basis of Disease, p. 583
Clinical Features
- Onset: Insidious
- Symptoms: Fatigue, weakness, weight loss, anorexia (hypermetabolism), dragging left upper quadrant sensation from splenomegaly, or acute left upper quadrant pain from splenic infarction
- Diagnosis confirmed by: Detection of BCR::ABL fusion gene via karyotype (Ph chromosome), FISH, or PCR
Response Definitions & Monitoring
| Response Type | Definition |
|---|---|
| Hematologic response | Normalization of CBC, no symptoms |
| Cytogenetic response (CCyR) | No Ph+ metaphases on bone marrow cytogenetics |
| Major Molecular Response (MMR) | ≥3-log reduction in BCR-ABL mRNA (BCR-ABL ≤0.1% on IS) |
| Deep Molecular Response (MR4/MR4.5) | ≥4-4.5 log reduction; prerequisite for treatment-free remission (TFR) attempts |
Treatment: Tyrosine Kinase Inhibitors (TKIs)
Six FDA-approved oral BCR::ABL1 TKIs (as of Harrison's 22E, 2025):
| Generation | Drug | Dose | Notable Toxicities |
|---|---|---|---|
| 1st | Imatinib (Gleevec) | 400 mg/day | GI, fluid retention, cytopenias |
| 2nd | Dasatinib (Sprycel) | 100 mg/day (1L); 140 mg/day (transformation) | Pleural/pericardial effusions, pulmonary hypertension, cytopenias |
| 2nd | Nilotinib (Tasigna) | See label | Cardiovascular (arterial occlusion), hyperglycemia, QTc prolongation |
| 2nd | Bosutinib (Bosulif) | See label | Diarrhea, hepatotoxicity, cytopenias |
| 3rd | Ponatinib (Iclusig) | See label | Hypertension, arterial/venous thrombosis (VEGFR inhibition) |
| 3rd/STAMP | Asciminib (Scemblix) | See label | Inhibits ABL myristoyl pocket (different MOA); active vs. T315I |
Key comparisons:
- Nilotinib: structurally similar to imatinib but 30× more potent
- Dasatinib: SRC + ABL inhibitor, 300× more potent than imatinib
- Bosutinib: SRC + ABL inhibitor, 30-50× more potent than imatinib
- Ponatinib + Asciminib: active against T315I "gatekeeper" mutation (resistance to 1st/2nd gen TKIs)
- Asciminib: STAMP inhibitor (Specifically Targeting the ABL Myristoyl Pocket) - distinct mechanism
Outcomes with imatinib (chronic-phase):
-
Complete cytogenetic remission (CCyR): ~60-70%
-
Major molecular response (MMR): ~50-60%
-
Overall survival >90%
-
2nd-generation TKIs achieve higher early response rates and less progression to advanced phases, but without a clear overall survival advantage
-
Harrison's Principles of Internal Medicine 22E, p. 882
Survival Impact of TKI Era
The figure below from the MD Anderson Cancer Center experience dramatically illustrates the TKI revolution:
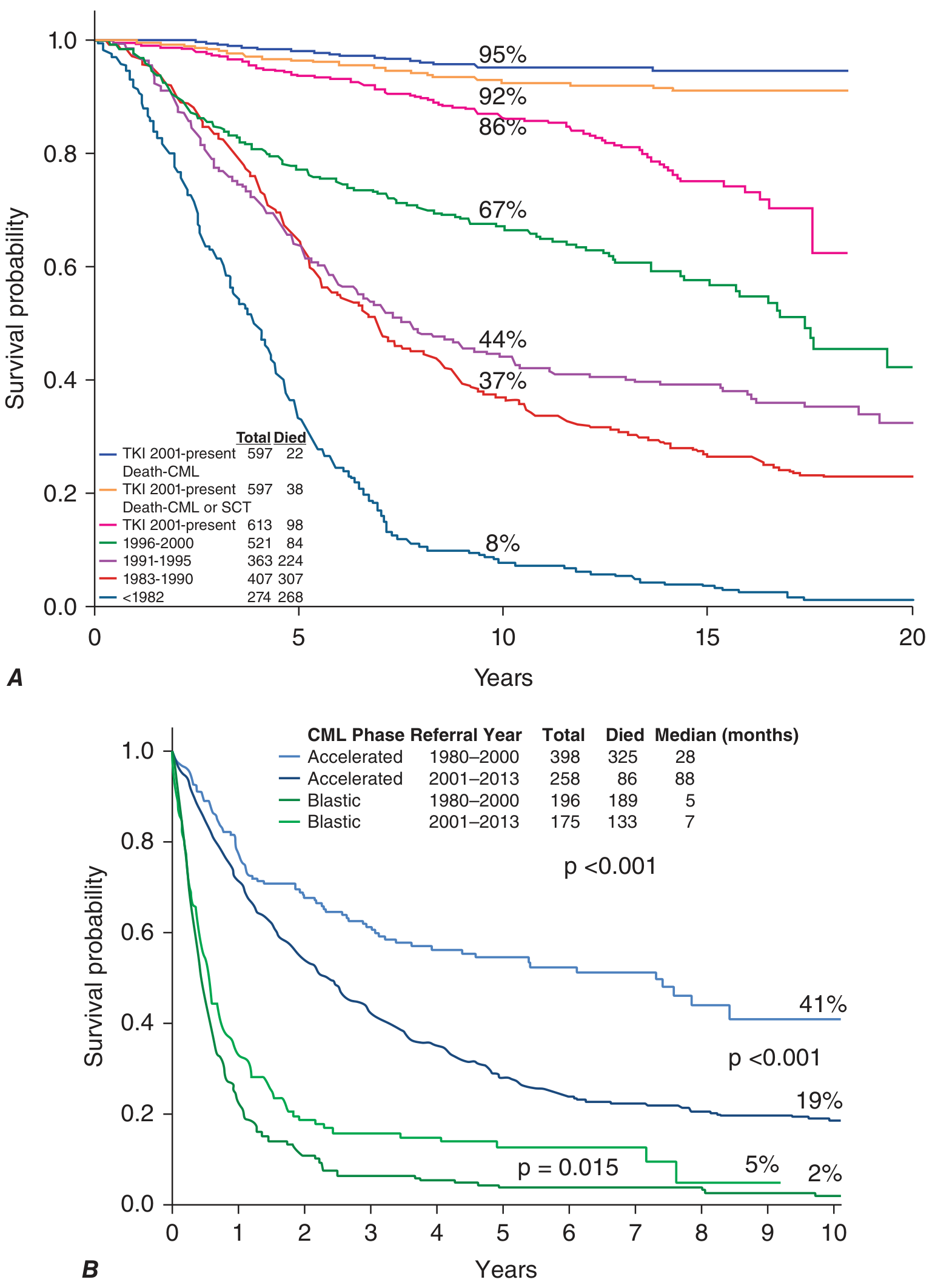
- Panel A (Chronic phase): TKI era (2001-present) achieved ~95% CML-specific survival at 20 years vs. only ~8% in the pre-1982 era
- Panel B (Accelerated/Blast phase): TKIs dramatically improved accelerated-phase survival (median 88 months vs. 28 months), but blast-phase benefit remains modest (median 7 vs. 5 months)
Treatment-Free Remission (TFR)
A major evolving goal in CML management. Patients who achieve deep molecular responses (MR4/MR4.5) on TKI therapy can attempt treatment discontinuation. Approximately 40-50% of eligible patients can sustain TFR. Those who lose response typically regain it rapidly on TKI restart.
Resistance & the T315I Mutation
The T315I "gatekeeper" mutation in ABL confers resistance to all 1st and 2nd generation TKIs (imatinib, dasatinib, nilotinib, bosutinib). Ponatinib and asciminib are the agents active against T315I disease. A 2024 systematic review (PMID: 39634261) compared ponatinib vs. asciminib in post-2nd-gen TKI failure.
Differential Diagnosis
| Feature | CML | Leukemoid Reaction |
|---|---|---|
| WBC | Often >100,000/µL | Usually <50,000/µL |
| Basophilia | Common | Absent |
| Ph chromosome / BCR-ABL | Present | Absent |
| LAP (leukocyte alkaline phosphatase) score | Low/absent | Elevated |
| Splenomegaly | Marked | Mild/absent |
Recent Evidence Note
A 2025 meta-analysis (PMID: 40317312) systematically reviewed patient-reported toxicity during TKI therapy in CML, highlighting that TKI side effects remain a significant quality-of-life burden even with effective disease control.
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease (10e), pp. 583-585
- Harrison's Principles of Internal Medicine 22E (2025), pp. 881-882
- Goldman-Cecil Medicine (International Ed.), pp. 1938-1940
This is a shared conversation. Sign in to Orris to start your own chat.