phenylalanine and tyrosine metabolism
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive information from the textbooks. Let me compile the full answer.
Phenylalanine and Tyrosine Metabolism
Overview
Phenylalanine is an essential amino acid; tyrosine is conditionally essential (nonessential only when adequate phenylalanine is present in the diet). Their metabolic pathways converge and yield both glucogenic and ketogenic products - ultimately fumarate and acetoacetate.
- Biochemistry, 8th ed - Lippincott Illustrated Reviews, p. 742
- Harper's Illustrated Biochemistry, 32nd Ed, p. 289
1. Phenylalanine → Tyrosine (The Gateway Reaction)
The first and rate-limiting step is the irreversible hydroxylation of phenylalanine to tyrosine:
Enzyme: Phenylalanine hydroxylase (PAH) - a mixed-function oxygenase
Cofactor: Tetrahydrobiopterin (BH4), synthesized from GTP
Mechanism:
Cofactor: Tetrahydrobiopterin (BH4), synthesized from GTP
Mechanism:
- One atom of O₂ incorporates as the hydroxyl group on tyrosine
- The other atom is reduced to H₂O
- BH4 is oxidized to dihydrobiopterin (BH2), then regenerated by NADPH-requiring dihydropteridine reductase
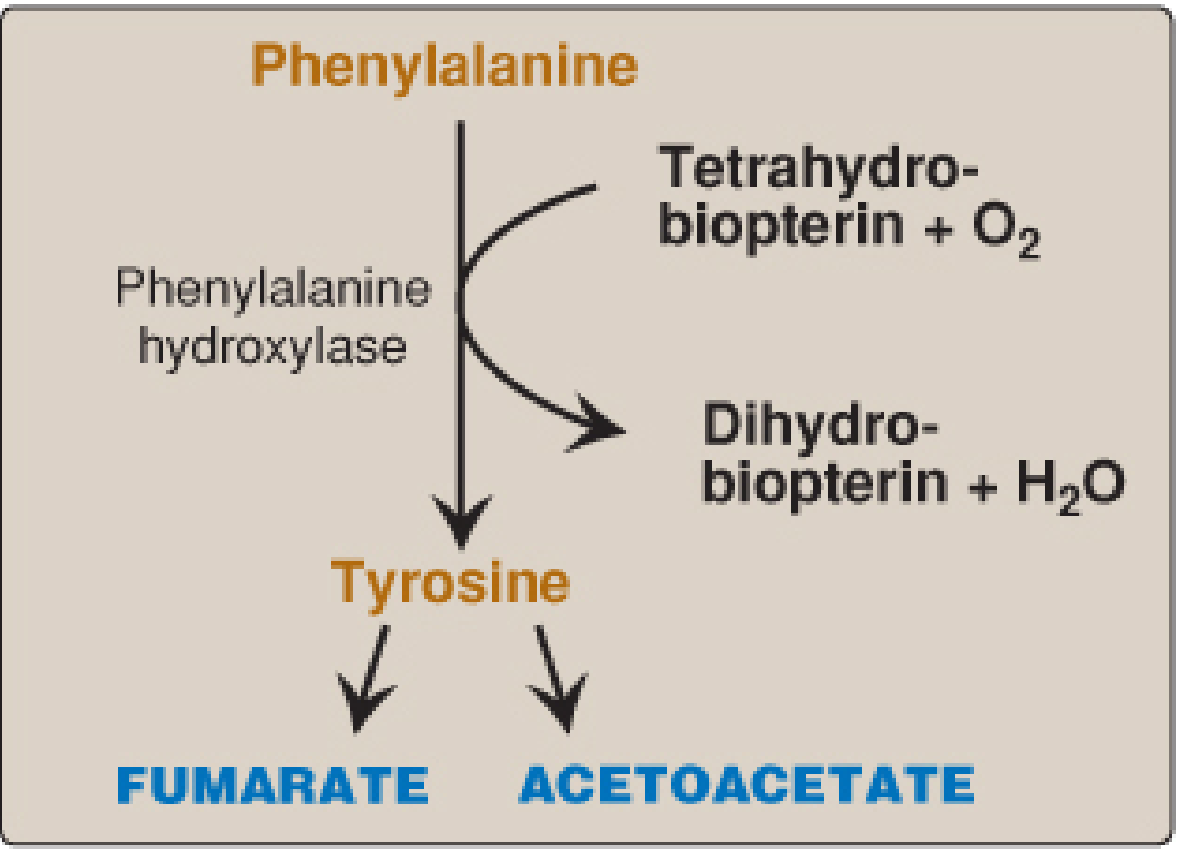
Fig. 20.7 from Lippincott Biochemistry
- Biochemistry, 8th ed - Lippincott, p. 742
2. Tyrosine Catabolism → Fumarate + Acetoacetate
Once tyrosine is formed, it undergoes further degradation through a multi-step pathway:
| Step | Enzyme | Product |
|---|---|---|
| 1 | Tyrosine aminotransferase | p-Hydroxyphenylpyruvate |
| 2 | 4-OH-Phenylpyruvate dioxygenase | Homogentisic acid |
| 3 | Homogentisic acid oxidase | Maleylacetoacetate |
| 4 | Maleylacetoacetate isomerase | Fumarylacetoacetate |
| 5 | Fumarylacetoacetate hydrolase | Fumarate + Acetoacetate |
This makes phenylalanine and tyrosine both glucogenic (via fumarate → oxaloacetate) and ketogenic (via acetoacetate).
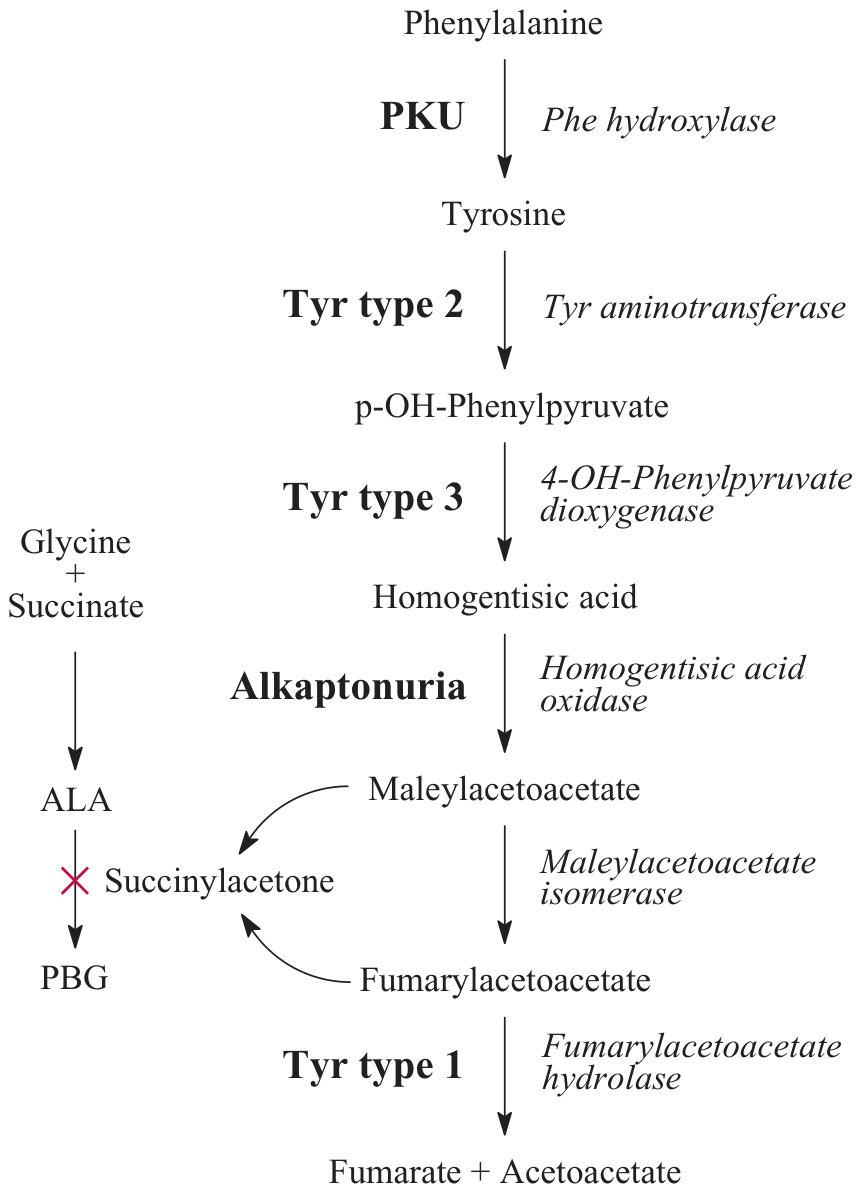
Phenylalanine/tyrosine degradation pathway with disease associations (Tietz Laboratory Medicine)
3. Biosynthetic Products of Tyrosine
Tyrosine is not only degraded - it is the precursor for several critical molecules:
A. Catecholamines (Neurons & Adrenal Medulla)
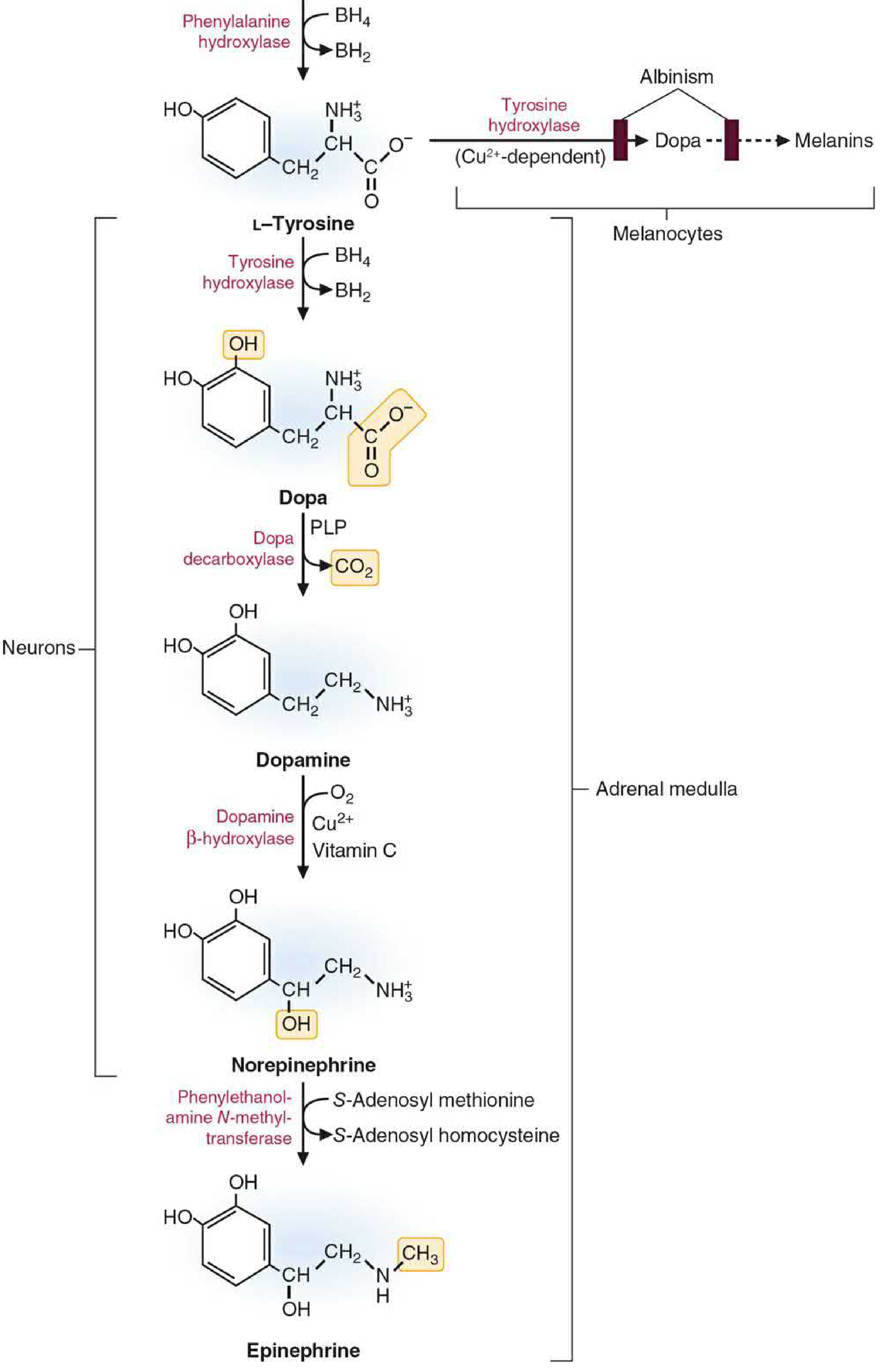
Basic Medical Biochemistry, Fig 46.4
Synthesis steps:
-
Tyrosine → L-DOPA
Enzyme: Tyrosine hydroxylase (rate-limiting step)
Cofactor: BH4 (same cofactor as PAH)
Note: Tyrosine hydroxylase is usually saturated, so dietary tyrosine levels do not readily alter catecholamine synthesis rate -
L-DOPA → Dopamine
Enzyme: DOPA decarboxylase (aromatic L-amino acid decarboxylase, AADC)
Cofactor: Pyridoxal phosphate (PLP/B6) -
Dopamine → Norepinephrine (in neurons & adrenal medulla)
Enzyme: Dopamine β-hydroxylase
Cofactors: O₂, Cu²⁺, Vitamin C (ascorbate) -
Norepinephrine → Epinephrine (in adrenal medulla only)
Enzyme: Phenylethanolamine N-methyltransferase (PNMT)
Methyl donor: S-adenosylmethionine (SAM)
- Basic Medical Biochemistry - A Clinical Approach 6e, p. 1632-1633
- Ganong's Review of Medical Physiology, p. 555-566
B. Melanin (Melanocytes)
In melanocytes, tyrosine is converted to DOPA by tyrosinase (a Cu²⁺-dependent enzyme - different from tyrosine hydroxylase), and DOPA is then oxidized further to produce melanin pigments. Deficiency of tyrosinase causes albinism.
C. Thyroid Hormones
Tyrosine residues in thyroglobulin are iodinated to form T3 (triiodothyronine) and T4 (thyroxine). This pathway operates within the thyroid follicular cells.
4. Summary Metabolic Map
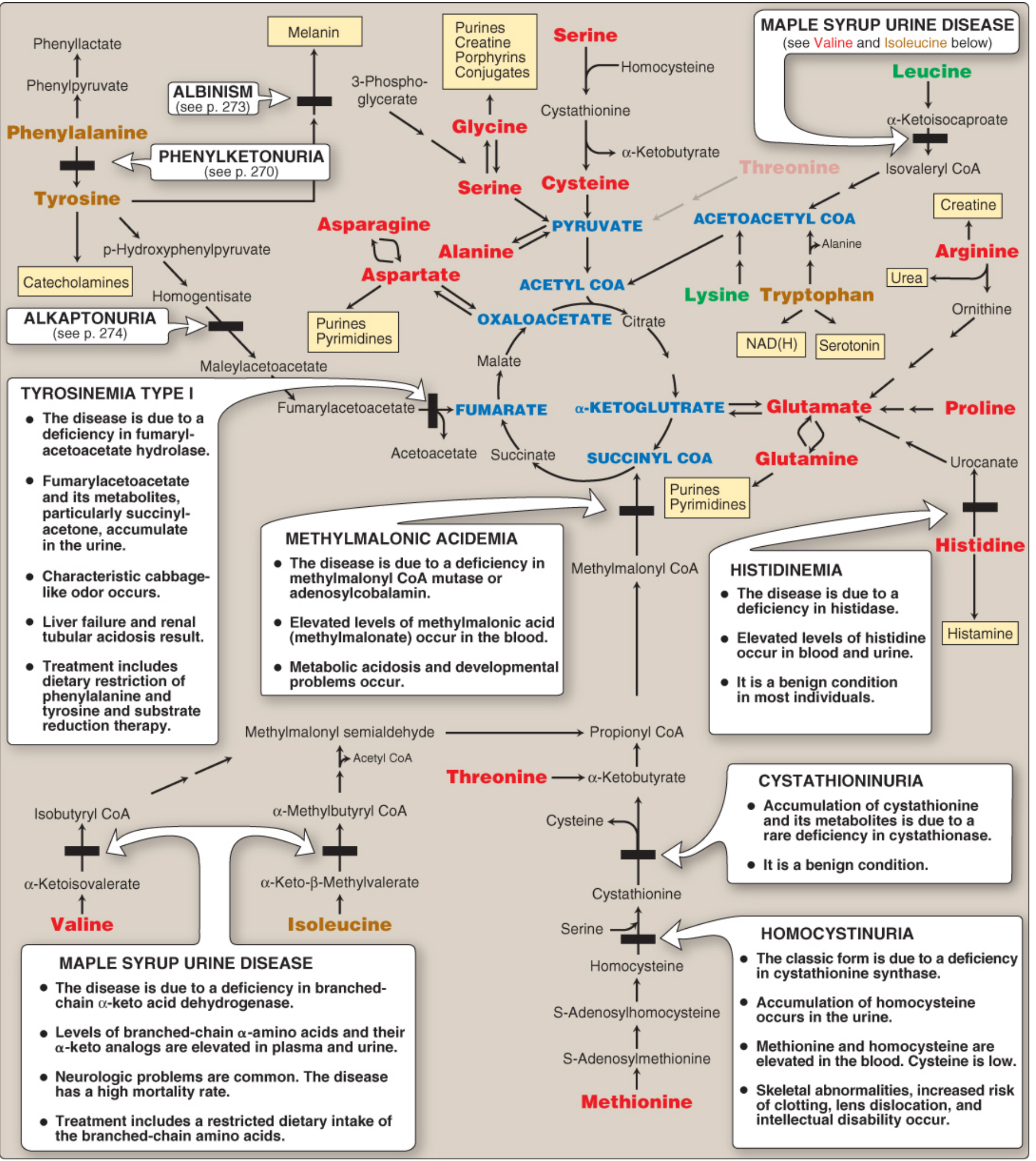
Fig. 20.15 from Lippincott Biochemistry - full amino acid metabolism map with associated disorders
5. Disorders of Phenylalanine & Tyrosine Metabolism
A. Phenylketonuria (PKU)
-
Defect: Loss-of-function mutations in PAH (100+ known mutations; autosomal recessive)
-
Incidence: ~1:15,000 - the most common inborn error of amino acid metabolism
-
Biochemistry: Hyperphenylalaninemia (10× normal); tyrosine becomes deficient; phenylalanine is shunted to phenylpyruvic acid and other metabolites excreted in urine
-
Clinical: Severe intellectual disability, seizures, fair hair/skin/eyes (reduced melanin), musty urine odor
-
Newborn screening: Blood phenylalanine measured after 24-48 hours of protein feeding (normal levels at birth because mother clears it via placenta)
-
Treatment:
- Phenylalanine-restricted diet (lifelong)
- Tyrosine supplementation (becomes essential)
- Avoid aspartame (contains phenylalanine)
- Must start within first 7-10 days of life to prevent cognitive damage
- Discontinuation even in adulthood leads to IQ deterioration
-
BH4-deficient variant: Rare; hyperphenylalaninemia plus deficient neurotransmitter synthesis (BH4 also needed for tyrosine hydroxylase and tryptophan hydroxylase). Dietary restriction alone is insufficient - requires BH4 + L-DOPA + 5-hydroxytryptophan supplementation
-
Maternal PKU: Women with PKU who become pregnant and resume normal diet expose the fetus to high phenylalanine - causes microcephaly, intellectual disability, and cardiac defects in the (often unaffected) offspring
-
Lippincott Biochemistry, p. 759-766
-
Emery's Elements of Medical Genetics and Genomics, p. 282
B. Tyrosinemia Type I (Hepatorenal Tyrosinemia)
- Defect: Fumarylacetoacetate hydrolase (FAH) - the last enzyme in tyrosine catabolism
- Key metabolite: Fumarylacetoacetate and its by-product succinylacetone accumulate; succinylacetone inhibits ALA dehydratase (heme synthesis)
- Clinical: Liver failure, renal tubular acidosis (Fanconi syndrome), characteristic cabbage-like urine odor
- Treatment: Dietary restriction of phenylalanine and tyrosine; nitisinone (NTBC) - inhibits 4-OH-phenylpyruvate dioxygenase to block production of toxic metabolites
C. Tyrosinemia Type II (Richner-Hanhart Syndrome)
- Defect: Tyrosine aminotransferase (first step of tyrosine catabolism, in the liver)
- Clinical: Corneal erosions (corneal crystals), painful palmoplantar keratosis, possible intellectual disability
D. Tyrosinemia Type III
- Defect: 4-OH-Phenylpyruvate dioxygenase
- Clinical: Mild - mainly neurological symptoms
E. Alkaptonuria
-
Defect: Homogentisic acid oxidase - autosomal recessive; the first inborn error of metabolism ever described (Garrod, 1902)
-
Clinical triad:
- Homogentisic aciduria - urine turns dark brown/black on standing (oxidation of HA)
- Ochronosis - black pigment deposits in cartilage, tendons, and collagenous tissue (ear cartilage, sclerae, intervertebral discs)
- Arthritis - severe degenerative arthritis of large joints (onset ~age 40)
-
Dark diaper staining may be the first sign in infants, but symptoms are typically absent until mid-life
-
Treatment: Dietary restriction of phenylalanine and tyrosine; nitisinone has also been explored
-
Lippincott Biochemistry, p. 771
F. Albinism
- Defect: Tyrosinase (Cu²⁺-dependent) - absent or defective; autosomal recessive (most common)
- Result: Absent or severely reduced melanin in skin, hair, and eyes
- Clinical: Hypopigmentation, photophobia, vision defects, increased risk of skin cancer
- Lippincott Biochemistry, p. 769
Quick Reference Summary
| Disorder | Enzyme Defect | Key Accumulation | Key Feature |
|---|---|---|---|
| PKU | Phenylalanine hydroxylase | Phenylalanine, phenylpyruvate | Intellectual disability; fair skin/hair |
| Tyrosinemia I | Fumarylacetoacetate hydrolase | Succinylacetone | Liver failure; cabbage odor |
| Tyrosinemia II | Tyrosine aminotransferase | Tyrosine | Corneal lesions, keratosis |
| Tyrosinemia III | 4-OH-phenylpyruvate dioxygenase | 4-OH-phenylpyruvate | Neurological |
| Alkaptonuria | Homogentisic acid oxidase | Homogentisic acid | Dark urine; ochronosis; arthritis |
| Albinism | Tyrosinase | N/A (reduced melanin) | No pigment; photophobia |
Key concept: Both phenylalanine and tyrosine are glucogenic AND ketogenic because their degradation produces both fumarate (enters TCA → gluconeogenesis) and acetoacetate (a ketone body). - Lippincott Biochemistry, p. 742
This is a shared conversation. Sign in to Orris to start your own chat.