Heme metabolism
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Loading Image
Loading Image
Reading File
I now have all the key information needed. Let me compile a comprehensive answer on heme metabolism.
Heme Metabolism
Heme is an iron-containing porphyrin ring that serves as a prosthetic group in hemoglobin, myoglobin, cytochrome P450 enzymes, and respiratory chain proteins. Its metabolism encompasses two distinct phases: synthesis (the Shemin pathway) and catabolism (the bilirubin pathway).
I. Heme Synthesis
Sites: ~85% occurs in erythroid precursor cells of the bone marrow; most of the remainder occurs in hepatocytes (primarily to make CYP enzymes). Mature RBCs lack mitochondria and cannot synthesize heme.
Compartmentalization: The first and last three steps occur in the mitochondria; the middle four steps occur in the cytosol.
The 8-Step Shemin Pathway
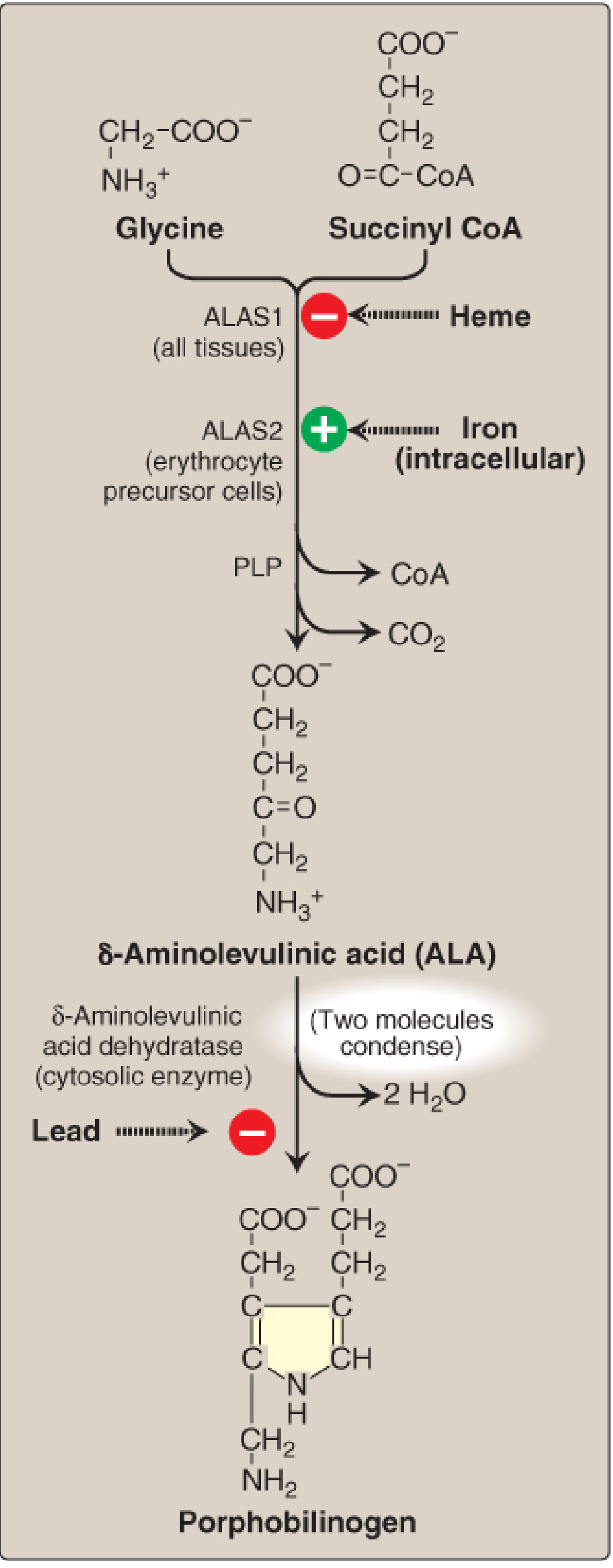
| Step | Location | Enzyme | Substrate → Product | Clinical Relevance |
|---|---|---|---|---|
| 1 | Mitochondria | ALA synthase (ALAS) | Glycine + Succinyl-CoA → δ-Aminolevulinic acid (ALA) | Rate-limiting, committed step; requires PLP (pyridoxal phosphate) |
| 2 | Cytosol | ALA dehydratase | 2 ALA → Porphobilinogen (PBG) | Inhibited by lead (Pb²⁺) |
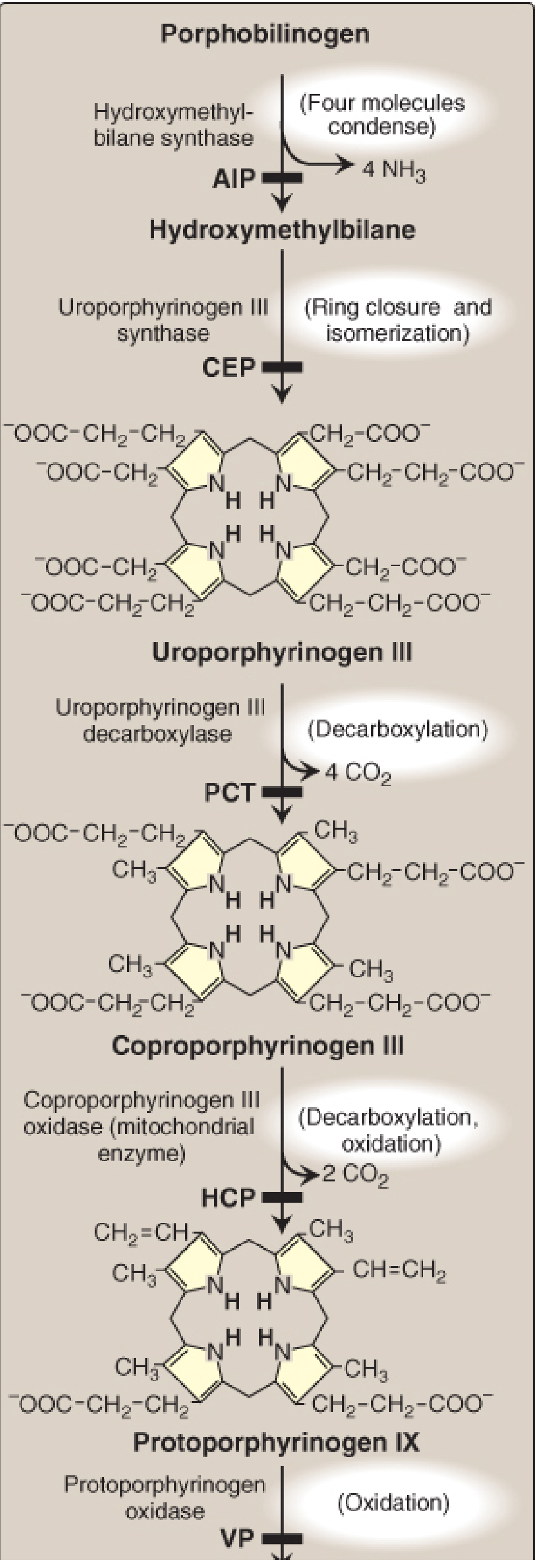
| 3 | Cytosol | Hydroxymethylbilane (HMB) synthase (= PBG deaminase) | 4 PBG → Hydroxymethylbilane | Deficiency → Acute Intermittent Porphyria (AIP) |
| 4 | Cytosol | Uroporphyrinogen III synthase | HMB → Uroporphyrinogen III | Deficiency → Congenital Erythropoietic Porphyria (CEP) |
| 5 | Cytosol | Uroporphyrinogen decarboxylase | Uroporphyrinogen III → Coproporphyrinogen III (4 CO₂ released) | Deficiency → Porphyria Cutanea Tarda (PCT) |
| 6 | Mitochondria | Coproporphyrinogen oxidase | Coproporphyrinogen III → Protoporphyrinogen IX | Deficiency → Hereditary Coproporphyria (HCP) |
| 7 | Mitochondria | Protoporphyrinogen oxidase | Protoporphyrinogen IX → Protoporphyrin IX | Deficiency → Variegate Porphyria (VP) |
| 8 | Mitochondria | Ferrochelatase (heme synthase) | Protoporphyrin IX + Fe²⁺ → Heme | Deficiency → Erythropoietic Protoporphyria (EPP) |
(Only type III porphyrins are physiologically important; all enzymes from step 6 onward are specific for type III.)
Regulation of ALA Synthase
There are two ALAS isoforms with distinct regulatory mechanisms:
ALAS1 (housekeeping - all tissues, especially liver):
- Feedback inhibition by heme (hemin): When heme accumulates, it represses ALAS1 gene transcription, accelerates mRNA degradation, and blocks mitochondrial import of the enzyme.
- Induction by drugs/xenobiotics: Drugs metabolized by CYP enzymes (e.g., barbiturates, rifampicin, phenytoin, trimethoprim) consume heme → free heme pool falls → ALAS1 is upregulated. This is the trigger for acute porphyria attacks.
ALAS2 (erythroid-specific - bone marrow):
- Regulated by intracellular iron availability (stimulated by iron)
- Loss-of-function mutations → X-linked sideroblastic anemia (XLSA)
- Gain-of-function mutations (exon 11) → X-linked protoporphyria (XLP)
"ALAS1 is regulated by heme; ALAS2 is regulated by iron." - Biochemistry, Lippincott Illustrated Reviews, 8th ed.
II. Heme Catabolism
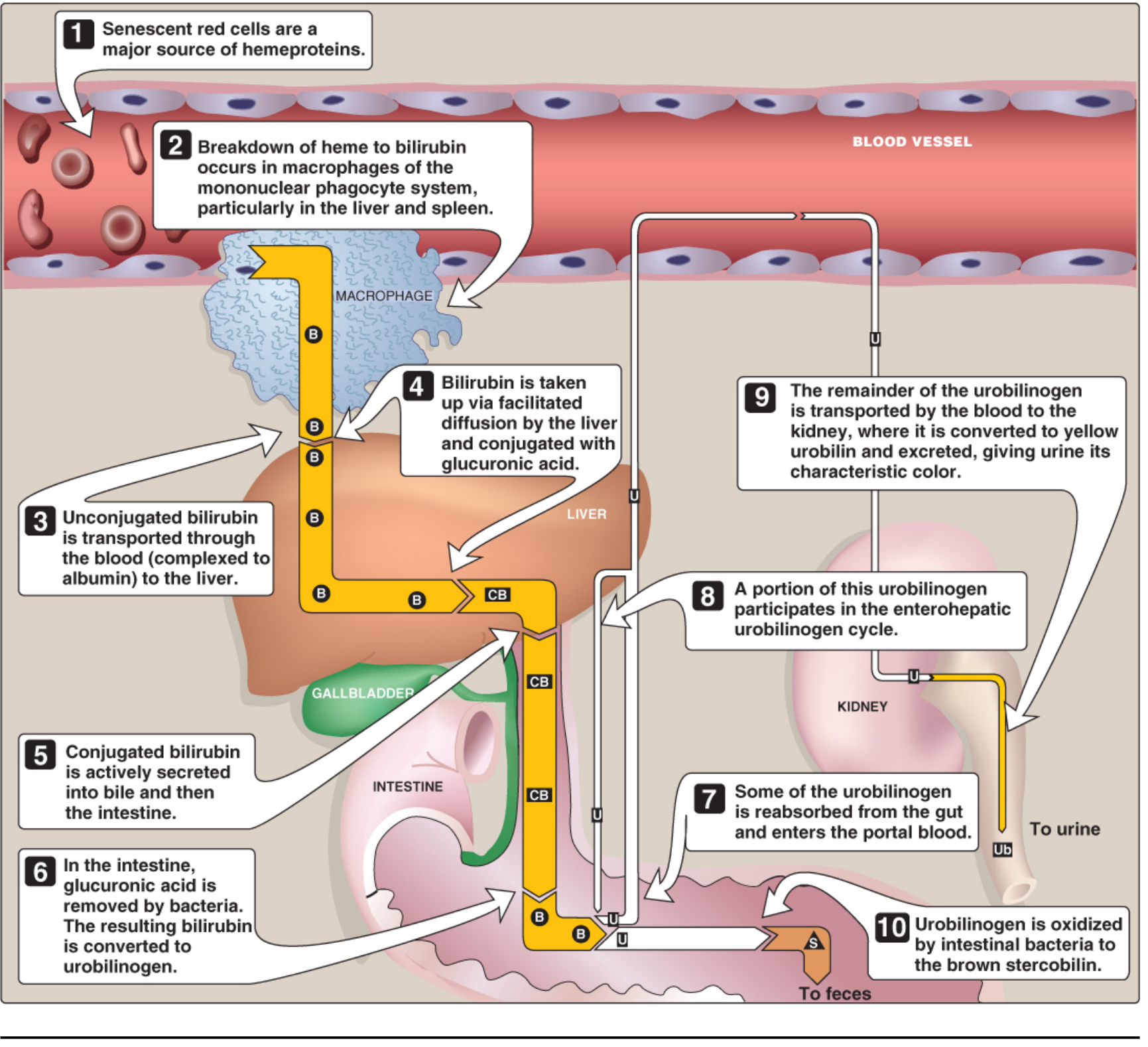
Step 1: Heme → Biliverdin → Bilirubin (in macrophages)
Senescent RBCs (lifespan ~120 days) are phagocytosed by macrophages of the mononuclear phagocyte system (mainly spleen, liver, bone marrow). Hemoglobin is split into globin (recycled as amino acids) and heme. The heme ring is then opened:
- Heme oxygenase cleaves the porphyrin ring, releasing:
- Fe²⁺ (recycled via transferrin)
- Carbon monoxide (CO) (only endogenous source of CO in humans)
- Biliverdin (a green, linear tetrapyrrole)
- Biliverdin reductase reduces biliverdin → bilirubin (yellow-orange)
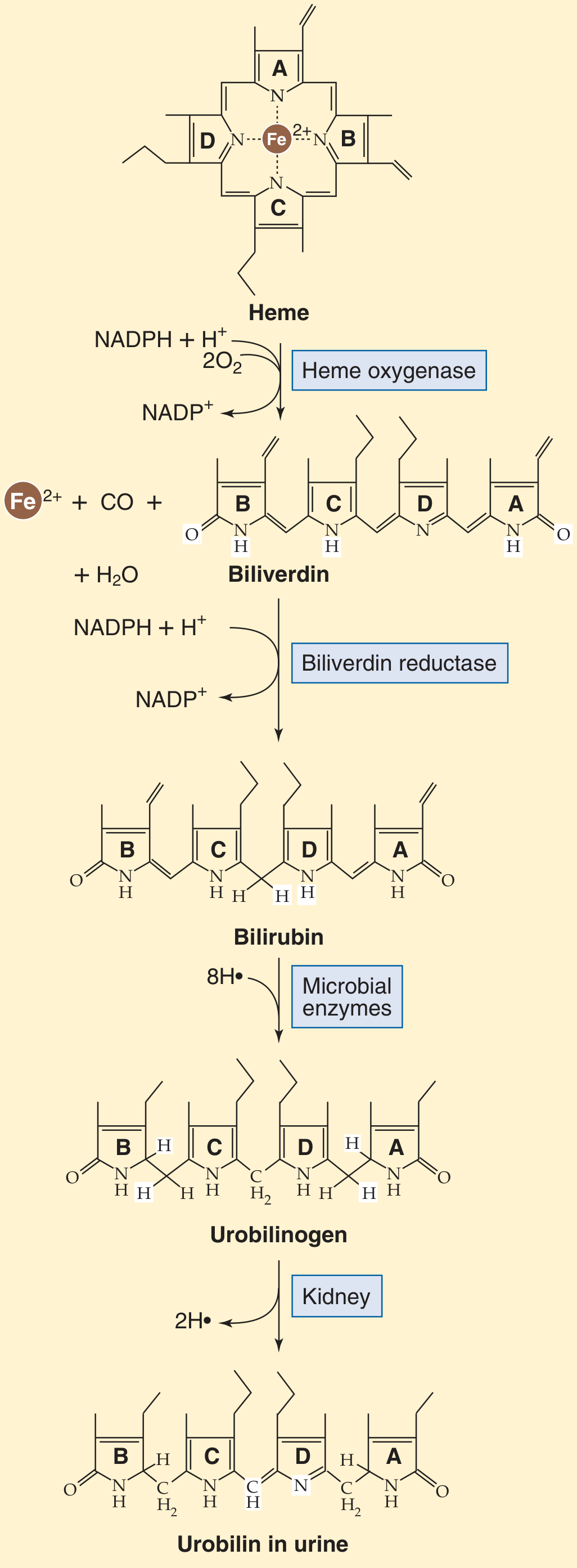
This newly formed bilirubin is unconjugated (indirect) bilirubin - lipophilic, water-insoluble, and potentially neurotoxic.
Step 2: Transport to the Liver
Unconjugated bilirubin binds non-covalently to albumin for transport through the blood (it is insoluble in plasma alone). Certain drugs (salicylates, sulfonamides) can displace bilirubin from albumin, raising the risk of kernicterus in neonates. In the hepatocyte, it enters by facilitated diffusion and binds intracellular proteins (particularly ligandin).
Step 3: Conjugation in the Liver
Hepatocytes conjugate bilirubin with glucuronic acid via bilirubin UDP-glucuronosyltransferase (UGT1A1), producing:
- Bilirubin monoglucuronide
- Bilirubin diglucuronide (conjugated / direct bilirubin - water-soluble)
Step 4: Secretion into Bile
Conjugated bilirubin is actively transported into bile canaliculi (energy-dependent, rate-limiting for the hepatic phase).
Step 5: Intestinal Fate
In the terminal ileum and colon, gut bacteria:
- Deconjugate (hydrolyze glucuronic acid) and reduce bilirubin → colorless urobilinogen
- Most urobilinogen is oxidized → brown stercobilin (color of feces)
- Some urobilinogen is reabsorbed → enterohepatic circulation; some escapes to kidneys → oxidized to yellow urobilin (color of urine)
III. Jaundice - Clinical Application
Jaundice appears at serum bilirubin ≥2-3 mg/dL (normal ≤1 mg/dL). Three types:
| Type | Cause | Elevated Fraction | Key Features |
|---|---|---|---|
| Hemolytic (pre-hepatic) | Excess RBC destruction (sickle cell, G6PD, pyruvate kinase deficiency) | Unconjugated | ↑ urobilinogen in urine/stool, no bilirubinuria |
| Hepatocellular | Hepatitis, cirrhosis, drugs | Both | ↑ conjugated + unconjugated; ↑ urine bilirubin |
| Obstructive (post-hepatic) | Gallstones, pancreatic cancer blocking bile duct | Conjugated | Pale stools, dark urine (bilirubinuria), pruritus |
Neonatal jaundice: Physiological in newborns due to immature UGT1A1 activity; unconjugated bilirubin can cross the blood-brain barrier → kernicterus. Treatment: phototherapy (converts bilirubin to water-soluble photoisomers).
Inherited conjugation defects:
- Crigler-Najjar syndrome type I: Complete absence of UGT1A1 → severe unconjugated hyperbilirubinemia; fatal without liver transplant
- Crigler-Najjar syndrome type II: Partial deficiency; responds to phenobarbital
- Gilbert syndrome: Mild UGT1A1 reduction (~30%); benign, triggers: fasting, stress
- Dubin-Johnson syndrome: Defect in canalicular transport of conjugated bilirubin → conjugated hyperbilirubinemia; benign
IV. Porphyrias - Enzyme Deficiency Disorders
Porphyrias result from deficiency (usually partial) in any enzyme of the Shemin pathway, causing accumulation of toxic intermediates:
| Porphyria | Deficient Enzyme | Type | Accumulates | Key Features |
|---|---|---|---|---|
| ADP (ALA dehydratase deficiency) | ALA dehydratase | Acute | ALA | Very rare; lead poisoning mimics this |
| AIP (Acute Intermittent Porphyria) | HMB-synthase (PBG deaminase) | Acute | ALA, PBG | Most common acute porphyria (5-10/100,000); abdominal pain, neurovisceral symptoms, dark urine |
| CEP (Congenital Erythropoietic Porphyria) | Uroporphyrinogen III synthase | Cutaneous | Uro I, Copro I | Severe photosensitivity, hemolysis, pink/red urine in infancy |
| PCT (Porphyria Cutanea Tarda) | Uroporphyrinogen decarboxylase | Cutaneous | Uroporphyrin | Most common porphyria overall; skin blisters/fragility in sun-exposed areas; associated with HCV, alcohol, iron overload |
| HCP (Hereditary Coproporphyria) | Coproporphyrinogen oxidase | Acute + Cutaneous | Coproporphyrin III | Can have both acute and skin symptoms |
| VP (Variegate Porphyria) | Protoporphyrinogen oxidase | Acute + Cutaneous | Proto + Copro | Common in South African whites; acute attacks + skin blisters |
| EPP (Erythropoietic Protoporphyria) | Ferrochelatase | Cutaneous | Protoporphyrin IX | Painful photosensitivity (rather than blisters), liver disease |
Triggers for acute porphyria attacks: Drugs (barbiturates, phenytoin, rifampicin, erythromycin, OCP progestins), fasting/low carbohydrate diet, alcohol, stress, infections, hormonal changes (especially progesterone - attacks more common in women post-puberty, premenstrually).
Treatment of acute attacks: Remove triggering agent; IV glucose (suppresses ALAS1 via carbohydrate loading); hematin/hemin (directly represses ALAS1 by feedback).
Summary Diagram
MITOCHONDRIA CYTOSOL MITOCHONDRIA
Glycine +
Succinyl-CoA
↓ ALAS1/ALAS2
ALA
↓ ALA dehydratase (inhibited by Pb)
PBG
↓ HMB synthase (AIP if deficient)
Hydroxymethylbilane
↓ UPG III synthase (CEP if deficient)
Uroporphyrinogen III
↓ UPG decarboxylase (PCT if deficient)
Coproporphyrinogen III
↓ Copro oxidase (HCP if deficient)
Protoporphyrinogen IX
↓ Proto oxidase (VP if deficient)
Protoporphyrin IX
↓ Ferrochelatase + Fe²⁺ (EPP if deficient)
HEME
Catabolism:
HEME → (Heme oxygenase) → Biliverdin + Fe²⁺ + CO
→ (Biliverdin reductase) → Unconjugated bilirubin (binds albumin)
→ (Liver: UGT1A1) → Conjugated bilirubin (water-soluble)
→ (Bile → Gut bacteria) → Urobilinogen → Stercobilin (feces, brown)
→ Urobilin (urine, yellow)
Key sources:
- Biochemistry, Lippincott Illustrated Reviews, 8th ed - detailed enzyme steps
- Harrison's Principles of Internal Medicine, 22E (2025) - clinical porphyrias
- Harper's Illustrated Biochemistry, 32nd ed - regulatory mechanisms
- Miller's Anesthesia, 10e - clinical liver/heme overview
- Medical Physiology (Boron & Boulpaep) - bilirubin secretion
This is a shared conversation. Sign in to Orris to start your own chat.