Complete corpus Callosal agenesis
Reading File
Finding Sources
Searching PubMed
"corpus callosum" AND "agenesis"
Searching the Web
complete corpus callosal agenesis clinical features diagnosis management 2024
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have comprehensive data from 5 authoritative textbooks plus recent PubMed evidence. Let me compile the full response.
Complete Corpus Callosal Agenesis (ACC)
1. Anatomy & Normal Development
The corpus callosum is the largest white matter commissure of the brain, containing approximately 200 million axons that connect the neocortex of both cerebral hemispheres. It develops between approximately 7 and 20 weeks' gestation, in parallel with the rest of the cerebrum. Its parts - in order of development - are the genu, body, splenium, and finally the rostrum.
A commissural plate differentiates within the lamina terminalis at day 39 of embryonic life. This plate acts as a bridge for axonal passage and provides a glial scaffold to guide decussating growth cones. The earliest callosal axons appear at 74 days, the genu and splenium are recognizable by 84 days, and adult morphology is achieved by 115 days.
2. Definition & Classification
The spectrum of corpus callosum developmental anomalies includes:
| Type | Description |
|---|---|
| Complete agenesis (ACC) | Total failure of the commissure to develop - no corpus callosum present |
| Partial agenesis (hypogenesis) | Shorter anterior-posterior length; missing segments (typically splenium and/or rostrum) |
| Hypoplasia | Normal length but abnormally thin |
| Hyperplasia | Thicker than expected |
In complete ACC, all callosal axons fail to cross the midline. These arrested axons form longitudinally oriented bundles of Probst, located medial to the lateral ventricles.
Note: Complete ACC is actually more common than partial agenesis. Partial ACC results from an insult to a previously normal corpus callosum.
3. Incidence & Epidemiology
-
Live births: ~1.8:10,000 prevalence
-
Children with developmental disabilities: 2%-3%
-
Patients with existing CNS anomaly: up to 47%
-
2.3% prevalence on CT scans in North America; 7%-9% in Japan
-
6.8% of fetuses with fetal alcohol syndrome have ACC
-
Creasy & Resnik's Maternal-Fetal Medicine, p. 399
-
Bradley and Daroff's Neurology in Clinical Practice, p. 1880
4. Pathogenesis
The pathogenesis centers on the commissural plate failure. If this plate is unavailable to guide axons across:
- Callosal fibers cannot cross the midline
- They are deflected posteriorly within their hemisphere of origin, forming the bundles of Probst
- Other axons pass into the anterior commissure (which may enlarge up to 4x its normal volume)
- Some aberrant fibers descend within the internal capsule with the corticospinal tract
The anterior commissure and hippocampal commissures are always well formed or enlarged in callosal agenesis - an important distinguishing feature.
5. Etiology & Associated Conditions
Approximately 30%-45% of cases have an identifiable cause:
Chromosomal (10%-17%)
- Deletions: del(4)(p16)
- Trisomies 8, 11, 13, 18, 21
- Duplications: dup(8)(p21p23)
Genetic Syndromes (20%-35%)
| Syndrome | Features |
|---|---|
| Aicardi syndrome | ACC + chorioretinal lacunae + vertebral anomalies + intellectual disability + myoclonic epilepsy; X-linked dominant (Xp22), almost exclusively in girls |
| Andermann syndrome | ACC + mental deficiency + peripheral neuropathy (autosomal recessive) |
| L1 syndrome | X-linked |
| Mowat-Wilson syndrome | - |
| Delleman (oculocerebrocutaneous) syndrome | ACC + orbital cysts + skin lesions |
| Alport syndrome | - |
| Oro-facial-digital syndrome | - |
Intrauterine Causes
- Cytomegalovirus (CMV) infection
- Toxoplasma gondii
- Fetal alcohol syndrome
- Other teratogens
Associated CNS Anomalies (85% of cases)
- Interhemispheric cysts
- Arachnoid cysts
- Chiari II malformation
- Dandy-Walker malformation
- Migration disorders (polymicrogyria, heterotopia, schizencephaly)
- Septo-optic dysplasia
- Lipoma of the interhemispheric fissure
- Colpocephaly
Complete ACC is more commonly associated with malformations of cortical development. Partial ACC is more often associated with posterior fossa abnormalities.
Non-CNS Anomalies (65% of cases)
- Craniofacial: macrocephaly, hypertelorism, broad/depressed nasal bridge, cleft lip/palate
- Skeletal: hand malformations, scoliosis
- Cardiac: VSD, PDA
- Ocular: coloboma, anophthalmia
- Genital: cryptorchidism
- Renal: hydronephrosis
6. Neuroimaging
MRI is the investigation of choice for diagnosing corpus callosal agenesis.
MRI Brain - Key Diagnostic Signs
Sagittal view:
- Complete absence of the corpus callosum
- No cingulate sulcus - vertically oriented sulci extend down to the third ventricle ("spoke-wheel" pattern, also called the sunburst sign in third trimester)
- No horizontally running cingulate sulcus
Axial view:
- Lateral ventricles in parallel orientation (instead of normal convergence)
- Colpocephaly: disproportionate dilation of the occipital horns (absence of splenium and posterior white matter hypoplasia)
- Superior elevation of the third ventricle between the bodies of lateral ventricles
Coronal view:
- Absent corpus callosum in the midline
- "Viking helmet" sign: widely spaced, upward-pointing anterior horns
- High-riding third ventricle
- Characteristic indentation on medial aspect of lateral ventricles by the bundles of Probst
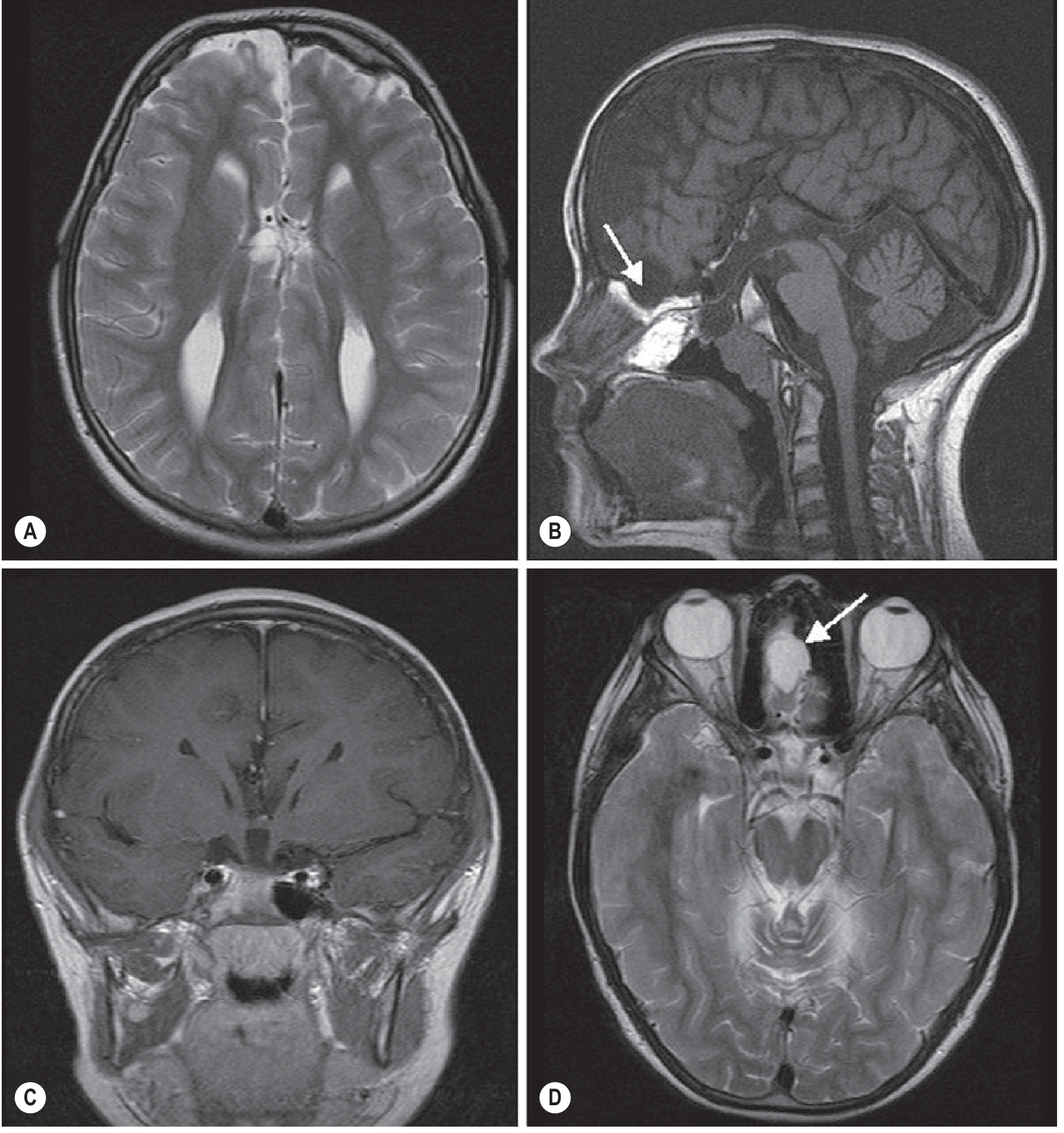
Callosal Agenesis MRI - Grainger & Allison's Diagnostic Radiology, p. 1979
Prenatal Ultrasound Signs
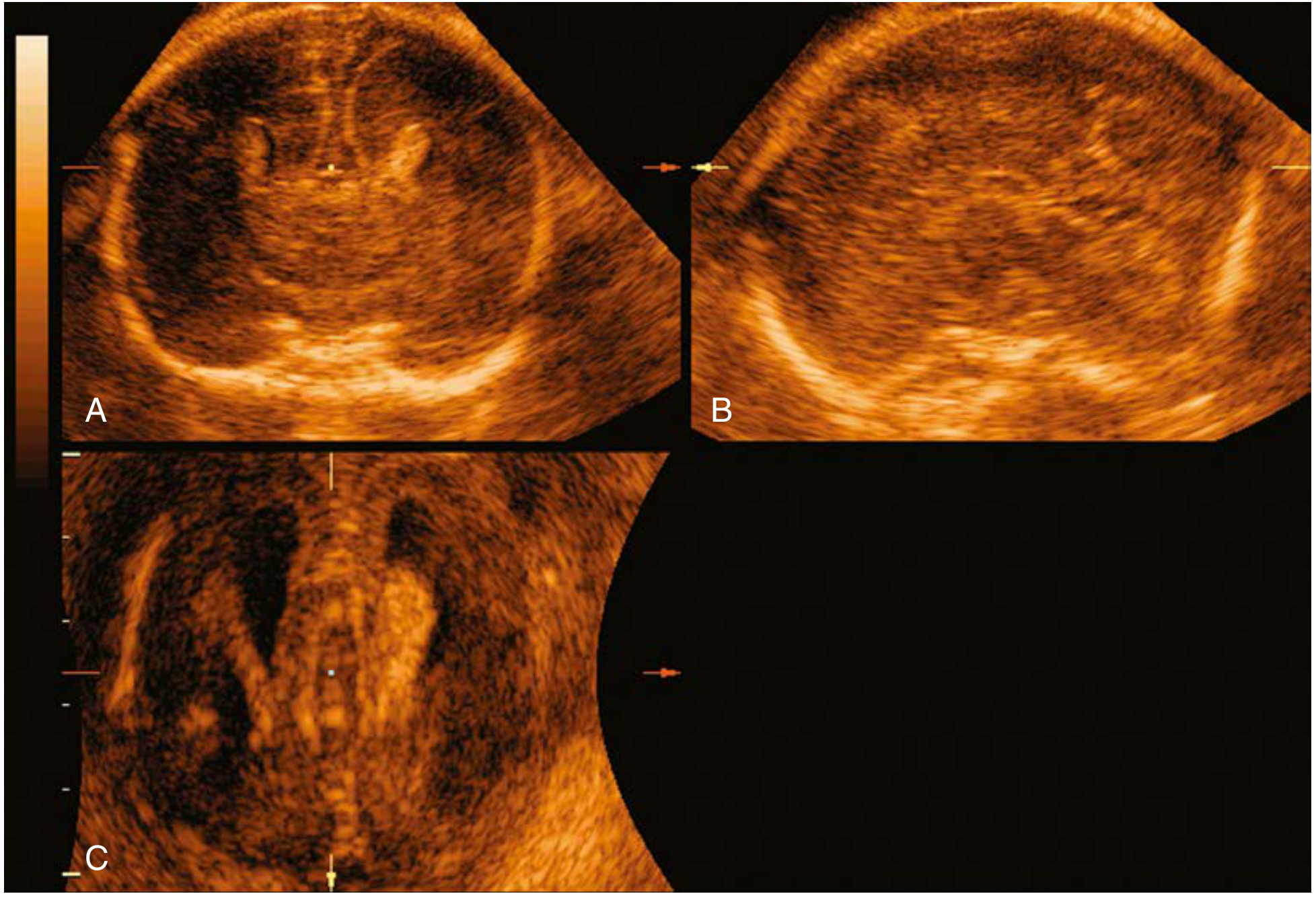
Prenatal ultrasound of ACC at 22 weeks - Creasy & Resnik's Maternal-Fetal Medicine, p. 399
Key antenatal ultrasound findings:
- Colpocephaly on axial section
- Absent cavum septi pellucidi
- Absent corpus callosum on median brain section
- Absent or abnormal pericallosal artery on median section with color Doppler
- Teardrop-shaped, parallel lateral ventricles on axial section
- Third-trimester "sunburst" sign - radial gyri and sulci on median surface
- "Viking helmet" sign - widely spaced, upward-pointing anterior horns on coronal section
7. Clinical Features
Clinical presentations range from completely asymptomatic to severe neurological disability:
Neurological
- Epilepsy: common, particularly in patients diagnosed early; seizures may relate more to associated focal cortical dysplasias than to callosal agenesis itself
- EEG: interhemispheric asynchrony, poor organization, multifocal spikes; asynchronous sleep spindles after 18 months is a useful clue
- Intellectual disability or learning disabilities
- Deficits in interhemispheric transfer of perceptual information for verbal expression
- Hypertelorism with exotropia and inability to converge
Neurodevelopmental / Cognitive (even in "normal IQ" individuals)
- Deficits in complex language and processing speed
- Visual-spatial reasoning difficulties
- Attention deficits
- Bimanual motor coordination problems
- Complex problem solving deficits
- Social functioning impairment - significant overlap with autism spectrum disorder (ASD) features
- Behavioral issues, emotional immaturity
Important note
Simple callosal agenesis may be clinically silent in children of normal intelligence; detailed neurological examination is required to uncover interhemispheric transfer deficits.
8. Differential Diagnosis
| Condition | Distinguishing Feature |
|---|---|
| Holoprosencephaly | Fused frontal horns, azygos anterior cerebral artery, absent interhemispheric fissure anteriorly |
| Septo-optic dysplasia | Optic nerve hypoplasia, absent septum pellucidum, pituitary insufficiency |
| Arachnoid cyst | Normal corpus callosum elsewhere; cyst displacing structures |
| Colpocephaly (primary) | CC may appear thinned but present |
| Hydrocephalus | Increased intraventricular pressure (absent in ACC) |
| Porencephaly / Schizencephaly | Cortical cleft communicating with ventricle |
The absence of septum pellucidum is distinguished from ACC by the presence of fused/communicating frontal horns and a discernible (even if thinned) corpus callosum.
9. Antenatal Management
Workup When Diagnosed Prenatally
- Detailed anatomic survey and fetal neurologic scan
- Fetal echocardiography (cardiac anomalies in 65%)
- Genetic counseling + karyotype; consider chromosomal microarray
- Fetal MRI - if ultrasound inconclusive, or to detect associated CNS anomalies (polymicrogyria, heterotopias, posterior fossa abnormalities) not visible on US
- Consider exome sequencing - a recent meta-analysis (PMID 37519216) showed significant diagnostic yield for prenatal ACC
- Offer pregnancy termination depending on family wishes
- In ongoing pregnancies: repeat scans in third trimester to reassess colpocephaly and/or hydrocephaly
Obstetric Management
- Delivery at a tertiary care facility with NICU and pediatric subspecialties
- Vaginal delivery is not contraindicated
- Cesarean delivery for obstetric indications only (or if associated Chiari II is present)
10. Postnatal/Neonatal Management
- MRI brain postnatally to confirm and characterize the anomaly
- Neurology consultation
- Neurosurgery consultation if interhemispheric cysts with hydrocephalus (may need shunting/drainage)
- Genetics consultation to exclude syndromic cause
- Neuropsychological evaluation - mandatory for all patients
- EEG if seizures suspected
- Anti-seizure medications for epilepsy (often medically intractable if associated with cortical dysplasia)
- Early intervention programs: speech therapy, occupational therapy, physiotherapy
- Developmental monitoring and school support
11. Prognosis
Outcomes span a very wide spectrum:
- Pooled rate of normal outcome: ~71.2% across all studies; 75.4% in MRI-confirmed studies
- Severe disability rate: 15.2% overall; 11.6% in MRI-confirmed studies
From a recent meta-analysis on isolated complete ACC:
| Outcome | Rate |
|---|---|
| Abnormal gross motor control | 4.40% |
| Abnormal fine motor control | 10.98% |
| Epilepsy | 6.80% |
| Abnormal cognitive status | 15.16% |
- Outcome is not clearly worse for complete vs. partial agenesis in isolation
- Individuals with ACC + other CNS anomalies are much more likely to have neurological symptoms
- "Asymptomatic" children may show emerging deficits as they age and face increasing cognitive demands
- Social/behavioral problems overlap with ASD in a significant proportion
- Early and continued intervention can improve outcomes
12. Key Associated Syndromes Summary
| Syndrome | Mode of Inheritance | Key Features |
|---|---|---|
| Aicardi | X-linked dominant (Xp22), lethal in males | ACC + chorioretinal lacunae + vertebral anomalies + myoclonic epilepsy; burst-suppression EEG |
| Andermann | AR | ACC + mental deficiency + peripheral neuropathy |
| L1 syndrome | X-linked | ACC + hydrocephalus + spastic paraplegia |
| Hirschsprung-ACC | Defective SMAD-interacting protein 1 (2q22-q23) | ACC + aganglionic megacolon |
| Mowat-Wilson | ZEB2 mutations | ACC + intellectual disability + Hirschsprung |
| Delleman (OCS) | - | ACC + orbital cysts + skin lesions + Dandy-Walker |
| Fetal alcohol syndrome | Teratogenic | ACC + dysmorphic features |
Recent Evidence
A 2024 meta-analysis (PMID 37519216, Ultrasound Obstet Gynecol) found significant diagnostic yield for exome sequencing in prenatal ACC - reinforcing the current recommendation to offer microarray/exome sequencing in all prenatally diagnosed ACC cases.
A 2023 review (PMID 37794643, Prenatal Diagn) provides updated counseling guidance for expecting parents with a prenatal ACC diagnosis, emphasizing the wide spectrum from normalcy to severe impairment.
A 2025 international Delphi consensus (PMID 40847729, Ultrasound Obstet Gynecol) has been published on prenatal evaluation, diagnosis and management of fetal corpus callosal abnormalities.
Sources: Bradley and Daroff's Neurology in Clinical Practice (pp. 1879-1880) | Creasy & Resnik's Maternal-Fetal Medicine (pp. 398-400) | Goldman-Cecil Medicine (Ch. 385) | Grainger & Allison's Diagnostic Radiology (pp. 1978-1980) | The Developing Human: Clinically Oriented Embryology (p. 1081)
This is a shared conversation. Sign in to Orris to start your own chat.