Waardenburgh syndrome
Waardenburg syndrome genetics clinical features
Waardenburg syndrome types genetics clinical features treatment 2024
Waardenburg Syndrome (WS)
- Scott-Brown's Otorhinolaryngology Head & Neck Surgery, p. 1338
- Andrews' Diseases of the Skin, p. 1484
Types and Genetics
| Type | Key Feature | Gene(s) | Inheritance |
|---|---|---|---|
| WS1 (Classic) | Dystopia canthorum present | PAX3 (2q35) | AD |
| WS2 | No dystopia canthorum; higher rate of hearing loss & heterochromia | MITF (3p14), SNAI2, SOX10 | AD |
| WS3 (Klein-Waardenburg) | Dystopia canthorum + limb abnormalities | PAX3 (2q35) | AD (>AR) |
| WS4 (Shah-Waardenburg) | WS2 features + Hirschsprung disease | EDNRB, EDN3, SOX10 | AD or AR |
- Dermatology 2-Volume Set 5e, Table 66.4, p. 1318-1319
- Andrews' Diseases of the Skin, p. 1493
Pathogenesis
Clinical Features
Pigmentary Abnormalities
- White forelock - the most frequent finding (20-60% of patients); usually white, though red/brown/black patches have been described
- Premature graying of scalp hair, eyebrows, and body hair (onset as early as teens)
- Leukoderma (hypopigmented skin patches) in ~15%, similar to piebaldism
- Heterochromia iridis (different colored eyes) or isohypochromia (pale blue eyes)
- Synophrys (medial eyebrow hyperplasia - eyebrows growing together)
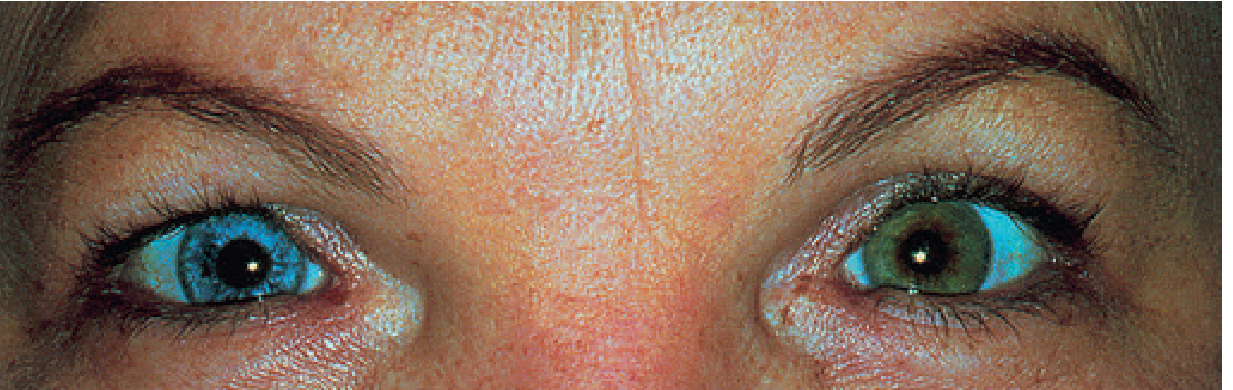
Dysmorphic Features
- Dystopia canthorum - increased distance between the inner canthi with normal interpupillary distance (present in WS1 and WS3; absent in WS2 and WS4)
- Broad nasal root
Hearing Loss
- Congenital sensorineural hearing loss - present in ~10-35% of WS1, more frequently in WS2
- Can be bilateral or unilateral, and ranges from mild to profound
- Caused by absence of melanocytes in the stria vascularis of the cochlea
Type-Specific Features
- WS3: Musculoskeletal anomalies - flexion contractures of elbows and fingers, hypoplasia of upper limb muscles, campodactyly
- WS4: Hirschsprung disease (congenital megacolon due to absent ganglion cells in the Auerbach plexus) - neural crest migration into the gut also requires EDN3/EDNRB and SOX10
Epidemiology
- Incidence: ~1 in 200,000 in the Netherlands; deafness from WS affects ~1 in 50,000 in the US
- Equal sex distribution; observed in all racial groups worldwide
- WS1 is the most common type (~55%), WS2 second most common (~40%)
Diagnosis
Management
- Hearing loss: Hearing aids or cochlear implants (particularly effective given the cochlear nerve is usually intact)
- Hirschsprung disease (WS4): Surgical resection of the aganglionic bowel segment
- Cosmetic concerns: Skin and hair pigmentation changes can be addressed cosmetically if desired
- Genetic counseling: For family planning and identifying at-risk relatives
- Multidisciplinary care: Audiology, ophthalmology, dermatology, and genetics
Key Differential Diagnoses
| Condition | Distinguishing feature |
|---|---|
| Piebaldism | No deafness, no eye findings, KIT gene mutations |
| Vitiligo | Acquired, not congenital |
| Tietz syndrome | MITF mutations; profound deafness + albinism but no heterochromia |
| Vogt-Koyanagi-Harada | Autoimmune, associated with uveitis |
- Dermatology 2-Volume Set 5e, p. 1318 (differential diagnosis table)
Search for recent research on gene therapy or novel treatments for WS
Waardenburg syndrome gene therapy treatment
PAX3 MITF SOX10 hearing loss melanocyte therapy
Waardenburg syndrome gene therapy novel treatment research 2024 2025
PMID: 33633356
cochlear gene therapy sensorineural hearing loss inner ear
cochlear implant gene therapy hearing loss children outcomes
PMID: 41085057
PMID: 38280389
PMID: 40690227
PMID: 40501308
Recent Research on Gene Therapy and Novel Treatments for Waardenburg Syndrome
Overview
1. The Key 2022 Review: Biological Therapeutics for WS
- Supportive techniques (hearing aids, cochlear implants, assistive listening devices) remain the only proven interventions to date
- Major preclinical progress has been achieved in gene delivery and stem cell replacement in animal models of genetic hearing loss
- The review maps out strategies and foreseeable barriers to implementing biological therapies for WS-related SNHL
- The central challenge: WS hearing loss involves absence of melanocytes from the stria vascularis (not hair cell loss), which is a distinct and less-studied target compared to hair cell gene therapies
2. Clinical-Stage Inner Ear Gene Therapy (Landmark Trials 2024-2026)
AAV1-hOTOF - The Lancet (2024)
- Intracochlear injection of AAV1 carrying the human OTOF transgene
- 5 of 6 children showed hearing recovery; ABR thresholds reduced by 40-57 dB from baseline (>95 dB)
- No dose-limiting toxicity or serious adverse events
- Speech perception improved in recovering participants
- Significance for WS: Demonstrates that intracochlear AAV delivery is safe and effective in children, validating the vector platform that could be adapted for WS
DB-OTO - New England Journal of Medicine (March 2026)
- Dual-AAV1 system delivering full-length OTOF cDNA via hair cell-specific promoter
- 75% of 12 children hit the primary endpoint (PTA ≤70 dB HL at week 24)
- 6 children could hear soft speech without any assistive devices
- 3 children achieved normal hearing sensitivity - a first in any congenital deafness gene therapy trial
- 67 adverse events, none causing trial discontinuation
- Significance for WS: This is the highest-quality evidence to date that inner ear gene therapy can restore near-normal hearing in congenital deafness. The dual-AAV approach also solves the large transgene packaging problem relevant to other hearing genes.
Gene Therapy vs. Cochlear Implant - JAMA Neurology (2025)
- First head-to-head comparison of OTOF gene therapy vs. cochlear implant
- GT patients outperformed CI patients on auditory/speech integration scores at 6 and 12 months
- GT patients showed better speech perception in noisy environments and better music perception (singing in-tune: 67% vs. 37% for CI)
- Shorter mismatch negativity latencies in GT group, suggesting more physiological auditory processing
- Implication: When WS-targeted gene therapies eventually enter trials, gene therapy may be preferred over cochlear implants if a functionally intact cochlear nerve is present (which it typically is in WS)
3. ATOH1 Gene Therapy for Hair Cell Regeneration
- Meta-analysis of 4 preclinical studies (52 rodents): ABR threshold reduction of -21 dB SPL (CI -40 to -2.5, p=0.027)
- Key upregulated genes: GFI1, PTPRQ, OTOF, USH2A, POU4F3 - all hair cell differentiation markers
- Still preclinical only; clinical trials needed
4. WS-Specific Research Directions (Preclinical)
| Strategy | Target | Status |
|---|---|---|
| AAV-mediated gene delivery of wild-type PAX3/MITF to stria vascularis | WS1/WS2 melanocyte loss | Preclinical (animal models) |
| Stem cell replacement - transplanting melanocyte progenitors derived from iPSCs into the cochlea | Stria vascularis repopulation | Early preclinical |
| CRISPR base editing to correct point mutations in PAX3, MITF | Mutation-specific correction | Conceptual/early research |
| Antisense oligonucleotides (ASOs) for splice-site mutations | WS2 (some MITF mutations) | Preclinical |
| Endothelin pathway modulation (EDN3/EDNRB agonists) | WS4 enteric neural crest defects | Early exploration |
Key Challenge Specific to WS
- Restore melanocyte colonization of the stria vascularis (a developmental window that may be prenatal), or
- Target embryonic/prenatal delivery to prevent melanocyte loss from occurring
5. Diagnostics: Expanding the Mutation Spectrum (2025)
- April 2025: Novel SOX10 frameshift mutation identified in a two-generation Chinese family via whole-exome sequencing, with reduced SOX10 expression confirmed in carriers - enabling preimplantation genetic testing
- 2026: A SOX10 truncating variant (p.Q59X) reported with co-occurrence of absent puberty and systemic lupus erythematosus (PMID: 41684881), broadening the known SOX10 phenotypic spectrum
Summary: Where Things Stand
| Domain | Current Status |
|---|---|
| WS-specific cure | None; no approved disease-modifying therapy |
| Hearing loss (WS) | Cochlear implants work well (cochlear nerve is intact) |
| Inner ear gene therapy (non-WS) | Phase I/II trials showing restoration of near-normal hearing in OTOF deafness (NEJM 2026) |
| WS gene delivery (preclinical) | Animal model studies ongoing; major barrier is stria vascularis biology |
| Stem cell therapy | Very early preclinical stage |
| Hirschsprung disease (WS4) | Surgical; no gene therapy trials |
| Prenatal/preimplantation genetics | Actively expanding; enabling families to prevent transmission |
Can u give me last year's 38th iap quiz question paper india
IAP 38th national pediatric quiz 2025 question paper India Indian Academy Pediatrics
IAP pedicon quiz 2025 38th edition question paper PDF India
https://www.scribd.com/document/878425174/38th-IAP-PAEDIATRI…
IAP 38th Undergraduate Pediatric Quiz (2025) - What I Found
- Divisional rounds were conducted across India (e.g., Cauvery Division at Indiana Hospital on 30 August 2025, SNIMS on 21 July 2025)
- The quiz document is uploaded on Scribd, with topics including:
- Portal vein anatomy
- Surfactant secretion
- Congenital conditions
- Core pediatric definitions
What You Can Do
-
Access the Scribd document directly (may need a subscription or free trial):
-
Contact your IAP State Branch - State coordinators who conducted divisional rounds often have the official question sets
-
IAP official website: iapindia.org - check under academic activities or contact the national quiz coordinator
-
Telegram/WhatsApp groups for IAP UG quiz preparation often circulate question papers among medical students - these are typically the fastest source