conditions when ALP goes high
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a thorough breakdown of all conditions that raise ALP, organized by source:
Elevated ALP - Causes by Source
ALP has isoenzymes in liver, bone, placenta, small intestine, and kidney. Elevated serum ALP almost always comes from the liver or bone. Knowing which organ is the source guides workup - a concurrent rise in GGT or 5'-nucleotidase (5'-NT) points to a hepatobiliary origin (not bone).
Hepatobiliary Causes
Cholestatic (ALP rises >4x normal)
- Primary biliary cholangitis (PBC) - one of the classic causes; anti-mitochondrial antibody (AMA) is positive
- Primary sclerosing cholangitis (PSC)
- Secondary sclerosing cholangitis
- Biliary obstruction - choledocholithiasis, stricture, bile duct carcinoma
- Drug-induced cholestasis - phenothiazines, anabolic steroids, oral contraceptives, erythromycin, chlorpromazine, and many others
- Intrahepatic cholestasis of pregnancy
- AIDS cholangiopathy - due to CMV or Cryptosporidium infection
Infiltrative liver disease (ALP often >4x normal)
- Hepatic metastases - ALP is the most sensitive hepatic chemistry marker for metastatic disease
- Primary liver tumors
- Amyloidosis with hepatic involvement
- Sarcoidosis of the liver
- Granulomatous infections - tuberculosis, fungal infections
- Langerhans cell histiocytosis
- Extramedullary hematopoiesis
Other hepatic
- Viral hepatitis - mild elevation (<3x) possible in any hepatitis
- Alcoholic hepatitis / steatohepatitis - rarely, values are greatly elevated
- Cirrhosis
- Congestive heart failure (hepatic congestion)
- Sepsis - can produce a cholestatic picture
- Liver transplant rejection
- Hodgkin's disease with liver involvement
- Diabetes mellitus (mild isolated elevation)
- Hyperthyroidism
Bone Causes (bone ALP elevated, GGT normal)
Bone ALP is produced by osteoblasts and reflects bone-forming activity. It rises whenever osteoblastic activity is increased.
| Condition | Notes |
|---|---|
| Paget's disease of bone | One of the most common causes of a markedly raised bone ALP in adults |
| Bone metastases (osteoblastic) | Prostate, breast cancer |
| Osteogenic sarcoma | |
| Healing fractures | Temporary rise |
| Osteomalacia / rickets | Secondary hyperparathyroidism drives osteoblast activity |
| Primary hyperparathyroidism | Osteitis fibrosa cystica in severe cases |
| Renal osteodystrophy | Secondary/tertiary hyperparathyroidism |
Physiological Elevations (non-pathological)
- Pregnancy (3rd trimester) - placental isoenzyme floods the bloodstream
- Childhood and adolescence - bone ALP is elevated due to rapid bone growth; values are up to twice adult levels
- Post-menopausal women - ALP rises gradually; a healthy 65-year-old woman has ALP ~50% higher than a healthy 30-year-old
- Blood groups O and B - after a fatty meal, intestinal ALP is released into blood; check ALP in the fasting state to avoid this
- Mild age-related rise in both sexes after age 60 (up to 1-1.5x normal)
Less Common / Miscellaneous
- Benign familial hyperphosphatasemia - persistent elevation of intestinal isoenzyme
- Congestive heart failure
- Renal cell carcinoma (ectopic ALP production)
- Hyperthyroidism (increased bone turnover)
Diagnostic Approach
The flowchart below (from Goldman-Cecil Medicine) guides workup of an isolated elevated ALP:
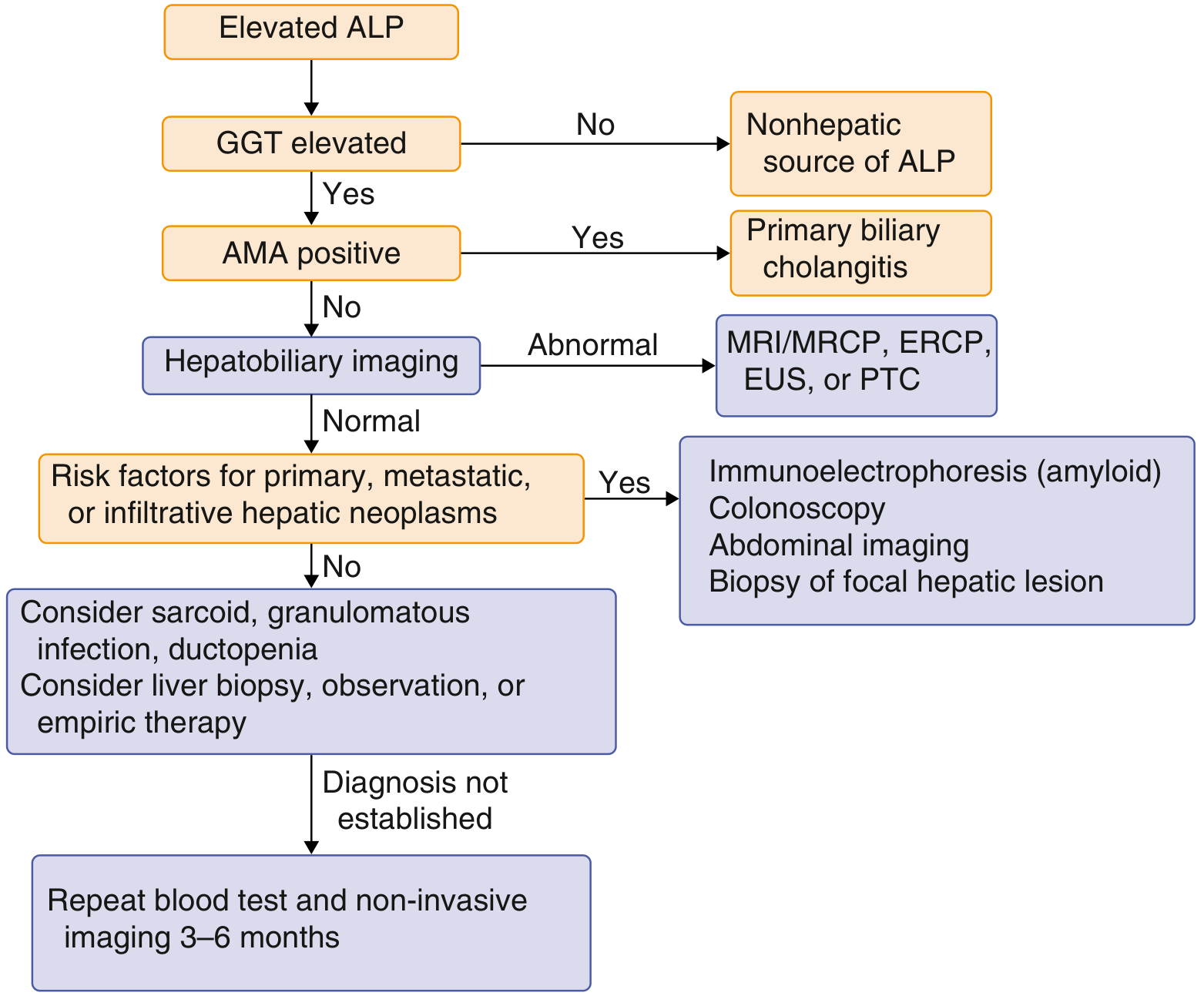
Key rule: If GGT is also elevated, the source is hepatobiliary. If GGT is normal, think bone, placenta, or intestine.
What LOWERS ALP (for contrast)
- Hypophosphatasia (inborn enzyme deficiency)
- Wilson's disease (copper displaces zinc cofactor)
- Malnutrition
- Hemolysis (falsely lowers measured value)
- Theophylline and estrogen therapy
Sources: Harrison's Principles of Internal Medicine 22E; Goldman-Cecil Medicine; Sleisenger & Fordtran's GI and Liver Disease; Quick Compendium of Clinical Pathology 5th ed.
what are the genetic traits should we corelate
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Here is a focused breakdown of the genetic traits and inherited disorders that correlate with ALP changes:
Genetic Correlations with ALP
1. Hypophosphatasia - ALPL Gene (LOW ALP)
- Gene: ALPL on chromosome 1p36 - encodes tissue-nonspecific ALP (TNSALP)
- Inheritance: Autosomal recessive (severe forms) or autosomal dominant (mild forms)
- Effect: Loss-of-function mutations - ALP is markedly LOW
- Substrates accumulate: inorganic pyrophosphate (PPi), phosphoethanolamine, pyridoxal-5'-phosphate
- Results in defective bone mineralization - mimics rickets/osteomalacia
- A low ALP + elevated vitamin B6 + bone disease = classic hypophosphatasia
- Tietz Textbook of Laboratory Medicine
2. Paget's Disease of Bone - SQSTM1 / TNFRSF11A (HIGH ALP)
- Genes: Mutations in SQSTM1 (sequestosome-1/p62) most common; also TNFRSF11A (RANK), VCP, ZNF687
- Inheritance: Autosomal dominant with variable penetrance; ~15-40% of cases are familial
- Effect: Unregulated osteoclast activity followed by chaotic osteoblast over-reaction - ALP can be 10-25x normal (one of the highest ALP elevations seen in any condition)
- Bone and liver ALP isoenzymes share a single gene and differ only by post-translational glycosylation, so fractionation is needed to confirm bone origin
- Goldman-Cecil Medicine; Henry's Clinical Diagnosis
3. Progressive Familial Intrahepatic Cholestasis (PFIC) - ATP8B1, ABCB11 (PARADOXICALLY NORMAL/LOW GGT despite HIGH ALP)
| Type | Gene | Protein | ALP | GGT |
|---|---|---|---|---|
| PFIC-1 | ATP8B1 | FIC1 (aminophospholipid flippase) | High | Normal/Low |
| PFIC-2 | ABCB11 | BSEP (bile salt export pump) | High | Normal/Low |
| PFIC-3 | ABCB4 | MDR3 (phospholipid transporter) | High | High |
- Key point: In PFIC types 1 and 2, serum ALP is elevated (cholestatic liver disease) but GGT is paradoxically normal - because reduced bile salt concentration keeps GGT anchored to the canalicular membrane rather than leaking into serum
- This is clinically important: the usual rule "elevated GGT = hepatic source of ALP" does not apply here
- Yamada's Textbook of Gastroenterology
4. Wilson's Disease - ATP7B Gene (LOW ALP)
- Gene: ATP7B on chromosome 13q14 - encodes a copper-transporting ATPase
- Inheritance: Autosomal recessive
- Effect: ALP is paradoxically LOW (or normal) despite fulminant hepatic failure
- Mechanism: excess copper displaces zinc (the ALP cofactor), inactivating the enzyme
- The triad of acute liver failure + hemolytic anemia + low ALP is highly characteristic of acute Wilson's disease
- Yamada's Textbook of Gastroenterology
5. Primary Biliary Cholangitis (PBC) - HLA & Immune Genes (HIGH ALP)
- Genetic susceptibility tied to HLA-DR8 and immune-regulatory loci (IL12A, IL12RB2, STAT4, IRF5)
- Autoimmune destruction of intrahepatic bile ducts causes cholestasis
- ALP is typically 4-10x normal and is often the first and only abnormal LFT
- Anti-mitochondrial antibody (AMA) against the pyruvate dehydrogenase complex (PDC-E2) is the hallmark
6. Primary Sclerosing Cholangitis (PSC) - HLA-B8/DR3 (HIGH ALP)
- Strong association with HLA-B8, HLA-DR3, HLA-DRw52a
- Also linked to IL2, IL2RA loci
- High association with IBD (especially ulcerative colitis) - shared genetic susceptibility
- ALP elevated as a dominant feature of the cholestatic pattern
7. Hereditary Hemochromatosis - HFE Gene (HIGH ALP - secondary)
- Gene: HFE - C282Y and H63D mutations (chromosome 6p21)
- Inheritance: Autosomal recessive
- Iron overload causes hepatic fibrosis/cirrhosis and eventually cholestasis
- ALP elevation is secondary to liver infiltration and damage
8. Alpha-1 Antitrypsin Deficiency - SERPINA1 Gene (HIGH ALP)
- Gene: SERPINA1 - PiZZ phenotype is most severe
- Inheritance: Autosomal codominant
- Misfolded Z-protein accumulates in hepatocytes causing progressive liver disease
- ALP elevation reflects intrahepatic cholestasis, especially in neonatal period
9. Blood Group Genes (ABO) - Physiological
- Blood groups O and B (glycosyltransferase gene variants on chromosome 9q34)
- Release intestinal ALP isoenzyme after fatty meals for up to 12 hours
- This is a normal physiological variant, not pathological - always check ALP in the fasting state
Summary Table
| Gene / Locus | Condition | ALP Direction | Mechanism |
|---|---|---|---|
| ALPL | Hypophosphatasia | ↓↓↓ | Loss-of-function - no ALP produced |
| SQSTM1, RANK | Paget's disease | ↑↑↑ | Osteoblast overactivity |
| ATP7B | Wilson's disease | ↓ | Copper displaces zinc cofactor |
| ATP8B1, ABCB11 | PFIC-1 & 2 | ↑ (GGT normal) | Cholestasis but bile salts retained |
| ABCB4 | PFIC-3 | ↑ (GGT high) | Phospholipid transport defect |
| HFE | Hemochromatosis | ↑ (secondary) | Iron-related liver damage |
| SERPINA1 | Alpha-1-AT deficiency | ↑ | Hepatocyte Z-protein accumulation |
| HLA-DR8, IL12A | PBC | ↑↑ | Autoimmune bile duct destruction |
| HLA-B8/DR3 | PSC | ↑↑ | Immune-mediated biliary stricturing |
| ABO blood group | Physiological variant | ↑ (mild, post-meal) | Intestinal ALP release |
Sources: Yamada's Textbook of Gastroenterology 7th ed; Tietz Textbook of Laboratory Medicine 7th ed; Goldman-Cecil Medicine; Harrison's Principles of Internal Medicine 22E; Rheumatology 2022
This is a shared conversation. Sign in to Orris to start your own chat.