In cardiac failure (HF), diuretics improve symptoms mainly by reducing congestion. Their effects on baroreceptors and osmoreceptors. Based on standard explanations from Guyton and Hall Textbook of Medical Physiology and Harrison’s Principles of Internal Medicine
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
Reading File
Reading File
I now have all the source material needed for a thorough, well-cited answer. Let me compose the full response.
Diuretics in Heart Failure: Effects on Baroreceptors and Osmoreceptors
Background - How HF Activates Neurohumoral Systems
In heart failure, reduced cardiac output causes arterial underfilling. Blood effectively "backs up" on the venous side while the arterial compartment is underfilled. This drop in arterial pressure is sensed by high-pressure baroreceptors, triggering a cascade that is both the disease's primary compensatory mechanism and a major driver of its symptoms.
According to Harrison's Principles of Internal Medicine (22nd ed.), Figure 264-7 (shown below), decreased cardiac output "unloads" the high-pressure baroreceptors in the left ventricle, carotid sinus, and aortic arch, resulting in:
- Reduced parasympathetic (afferent inhibitory) tone
- Generalized increase in efferent sympathetic tone
- Non-osmotic release of arginine vasopressin (AVP/ADH) from the pituitary
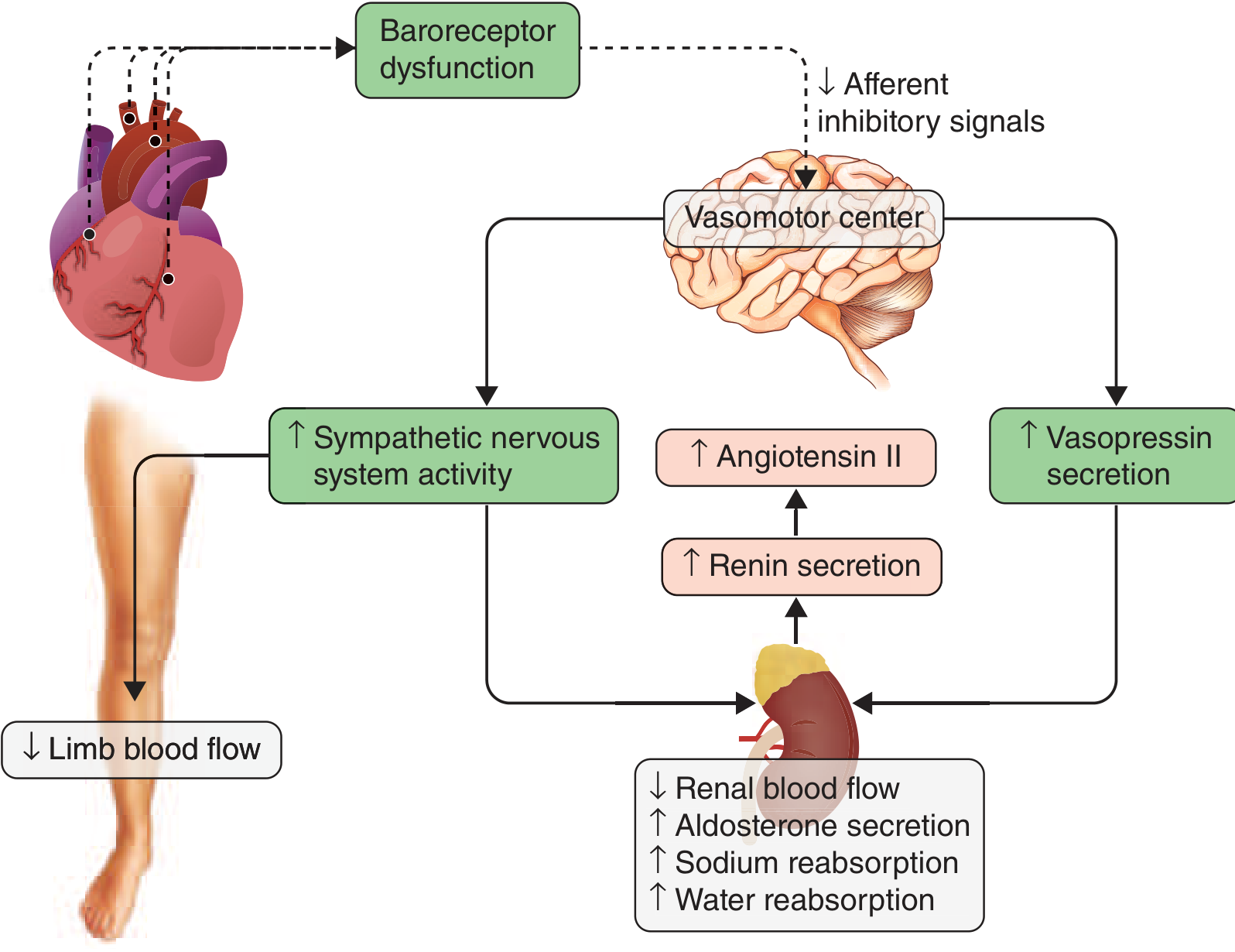
Harrison's Fig. 264-7 - Neurohormonal activation cascade in heart failure
1. Baroreceptors in HF - The Core Problem
Baroreceptor Resetting (Katzung, 16th ed., p. 334):
"The baroreceptor reflex appears to be reset, with a lower sensitivity to arterial pressure, in patients with heart failure. As a result, baroreceptor sensory input to the vasomotor center is reduced even at normal pressures; sympathetic outflow is increased, and parasympathetic outflow is decreased."
This means:
- Even when MAP appears "normal," the baroreceptors fire less than they should
- The vasomotor center receives reduced inhibitory afferent signals
- The result: chronically elevated sympathetic tone driving tachycardia, vasoconstriction (increasing afterload), and renin release
Costanzo Physiology (7th ed., Box 4.5) explains the mechanism clearly: decreased arterial pressure (Pa) causes the baroreceptor reflex to fire -- decreased Pa activates baroreceptors, increasing sympathetic outflow to the heart and vessels (raises HR, produces cutaneous vasoconstriction) while decreasing parasympathetic outflow. This is why the classic HF patient has tachycardia and cold, clammy skin.
Below is the Katzung flow diagram showing how reduced carotid sinus firing (baroreceptor silencing) feeds the vicious cycle of preload and afterload increase:
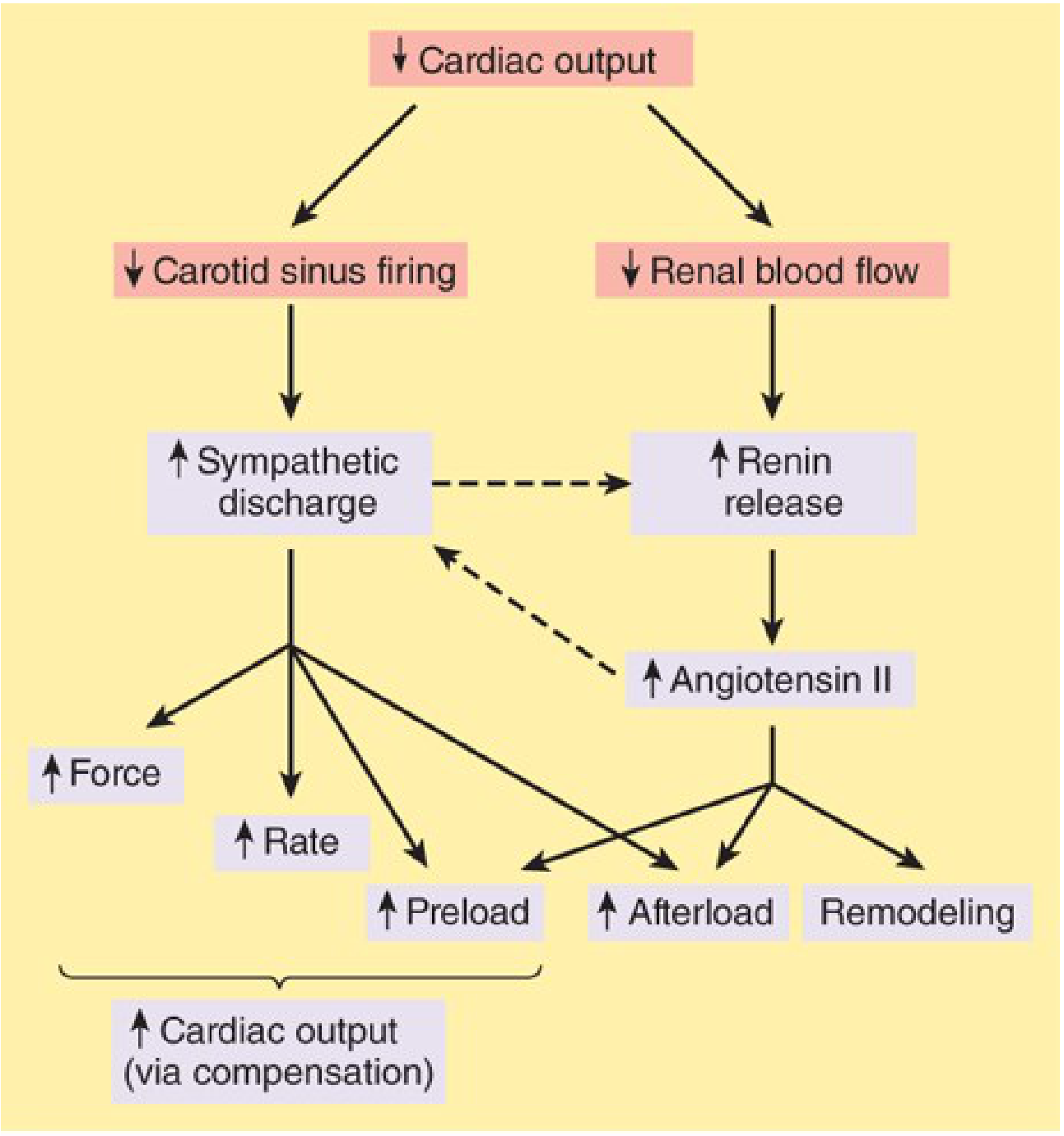
Katzung Fig. 13-2 - Baroreceptor-mediated compensatory responses in HF
Additionally, Harrison's notes that over time, "unchecked compensatory responses lead to excessive vasoconstriction and volume retention, electrolyte and renal abnormalities, baroreceptor dysfunction, direct myocardial toxicity, and cardiac arrhythmias." Aldosterone worsens this - Katzung notes that aldosterone "may also cause myocardial and vascular fibrosis and baroreceptor dysfunction in addition to its renal effects," which is one reason why mineralocorticoid antagonists (spironolactone, eplerenone) improve outcomes.
2. Vasopressin (ADH) and Osmoreceptors in HF - Non-Osmotic Release
Normally, vasopressin (ADH) is released by hypothalamic osmoreceptors in response to rising plasma osmolality (hyperosmolality). This is the classic osmoreceptor-driven pathway. In HF, a second, overriding pathway operates.
Harrison's (22nd ed.) explicitly states that baroreceptor unloading in HF causes non-osmotic release of AVP from the posterior pituitary. This is separate from, and can override, normal osmoreceptor inhibition. The consequences are:
- Vasopressin acts as a potent vasoconstrictor (V1 receptors), worsening afterload
- Vasopressin causes free water retention at the collecting duct (V2 receptors), diluting serum sodium and perpetuating volume overload
This is the physiological basis for dilutional hyponatremia in advanced HF - the plasma osmolality may be low (which would normally suppress osmoreceptors and shut off ADH), but the baroreceptor override keeps ADH elevated anyway.
Katzung's (Chapter 15 - HEART FAILURE section, p. 421) further notes:
"When cardiac output is reduced by heart failure, the resultant changes in blood pressure and blood flow to the kidney are sensed as hypovolemia and lead to renal retention of salt and water."
The kidney's response mirrors the baroreceptor interpretation: both systems read low cardiac output as "volume depletion" and respond with retention - appropriate in true hypovolemia, maladaptive in HF.
3. How Diuretics Reverse These Neurohumoral Changes
Primary mechanism - reducing venous congestion and preload:
Katzung states: "They reduce salt and water retention, reduce edema through renal effects, and reduce heart failure symptoms. They have no direct effect on cardiac contractility; their major mechanism of hemodynamic action in heart failure is to reduce venous pressure and ventricular preload."
When diuretics successfully reduce volume overload, they work "upstream" on the entire neurohumoral cascade:
A. Effect on Baroreceptors
By reducing total circulating volume and venous congestion, effective diuresis:
- Offloads the pulmonary and systemic venous beds
- Allows the heart to work at a more efficient fiber length (smaller end-diastolic volume)
- Improves forward cardiac output (to a degree)
- As cardiac output improves, arterial baroreceptors are better loaded, afferent baroreceptor firing increases, and the vasomotor center receives more inhibitory input
- Sympathetic outflow decreases, RAAS activity is partially suppressed
Frameworks for Internal Medicine puts it directly:
"Diuretics often improve hyponatremia associated with heart failure by optimizing preload, which improves cardiac output and effective arterial blood volume, thereby inhibiting baroreceptor-mediated vasopressin release."
So the baroreceptor effect of diuretics is indirect - by improving the hemodynamic state, they allow baroreceptors to recover toward normal sensitivity and reduce pathological sympathetic drive.
B. Effect on Osmoreceptors / ADH Pathway
In HF, the ADH elevation is primarily baroreceptor-driven (non-osmotic), not osmoreceptor-driven. Therefore:
- Diuretics do not act on osmoreceptors directly
- Diuretics restore baroreceptor loading (mechanism above) → reduces non-osmotic AVP drive
- With less AVP, free water retention decreases → plasma Na+ tends to normalize
- Serum osmolality rises back toward normal, and now osmoreceptor feedback can again appropriately suppress AVP
This is why Frameworks notes diuretics "often improve hyponatremia associated with heart failure" - they break the baroreceptor → non-osmotic ADH → water retention loop.
Caution with thiazides: Frameworks also notes that "diuretic therapy, particularly thiazide agents, may worsen hyponatremia in some patients." This is because thiazides impair the kidney's diluting capacity (they block NaCl reabsorption in the diluting segment, the distal tubule), so even with reduced ADH, the kidney cannot excrete a dilute urine effectively.
C. The Vaptan Connection
Katzung (p. 421) notes the growing interest in vaptans (vasopressin V2 receptor antagonists) in HF - these directly block the osmoreceptor/ADH axis at the collecting duct. This is conceptually the pharmacological complement to what diuretics achieve indirectly.
4. The Risks - When Diuretics Overshoot
Katzung (p. 421) warns:
"In treating the heart failure patient with diuretics, it must always be remembered that cardiac output in these patients is being maintained in part by high filling pressures. Therefore, excessive use of diuretics may diminish venous return and further impair cardiac output."
If ventricular filling pressure drops below ~15 mmHg in the left ventricle:
- Frank-Starling reserve is exhausted
- Cardiac output falls further
- Baroreceptors are further unloaded - worsening sympathetic and ADH activation
- Osmoreceptors may now also activate (if hyponatremia is corrected too fast, or if intravascular dehydration raises osmolality) → paradoxically increases ADH → perpetuating a new form of fluid imbalance
This creates the "over-diuresis paradox" - too much diuresis recreates the baroreceptor-mediated neurohormonal storm it was meant to suppress.
Summary Table
| Mechanism | In Untreated HF | After Diuretic Therapy |
|---|---|---|
| Baroreceptor sensitivity | Reset/blunted; fires less at normal pressures | Partially restored as cardiac output improves |
| Sympathetic outflow | Increased (tachycardia, vasoconstriction) | Reduced |
| ADH release | Non-osmotically elevated (baroreceptor-driven) | Reduced via improved baroreceptor loading |
| Osmoreceptor role | Overridden by non-osmotic (baroreceptor) ADH drive | Resumes physiological control as hemodynamics normalize |
| RAAS | Activated | Partially suppressed |
| Plasma Na+ | Often low (dilutional hyponatremia) | Tends to normalize (except with thiazides) |
| Risk of excess | - | Baroreceptor re-activation + osmoreceptor stimulation if over-diuresis occurs |
Sources:
- Harrison's Principles of Internal Medicine, 22nd ed. - Fig. 264-7 caption; Chapter 264 Mechanisms of Disease Progression
- Katzung's Basic and Clinical Pharmacology, 16th ed. - Chapter 13 (Pathophysiology of HF, Diuretics in HF); Chapter 15 (Heart Failure, p. 421)
- Costanzo Physiology, 7th ed. - Box 4.5 Clinical Physiology: Heart Failure
- Frameworks for Internal Medicine - Extrarenal causes of hypervolemic hyponatremia; baroreceptor-mediated vasopressin section
Guyton & Hall (Cardiac Failure and Body Fluid Regulation chapters Explained in concise manner
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed from Guyton & Hall directly. Let me compose the concise, well-structured answer.
Diuretics in Heart Failure - Guyton & Hall Perspective (Concise)
Source: Guyton and Hall Textbook of Medical Physiology - Chapters on Cardiac Failure (Ch. 22) and Body Fluid Regulation (Ch. 19, 29, 30)
1. What Happens in HF: The Baroreceptor Reflex is Triggered First
When the heart is suddenly damaged (e.g., myocardial infarction), cardiac output falls and blood dams up in the veins. Guyton & Hall (Ch. 22) describe the immediate response:
"The best known of these is the baroreceptor reflex, which is activated by diminished arterial pressure. The chemoreceptor reflex, the CNS ischemic response, and even reflexes that originate in the damaged heart also likely contribute to activation of the sympathetic nervous system."
Baroreceptors are spray-type stretch receptors in the walls of the carotid sinus and aortic arch. When arterial pressure drops:
- Baroreceptor firing rate falls
- Inhibitory afferent signals to the vasomotor center (nucleus tractus solitarius) decrease
- Sympathetic outflow surges; parasympathetic (vagal) tone is withdrawn
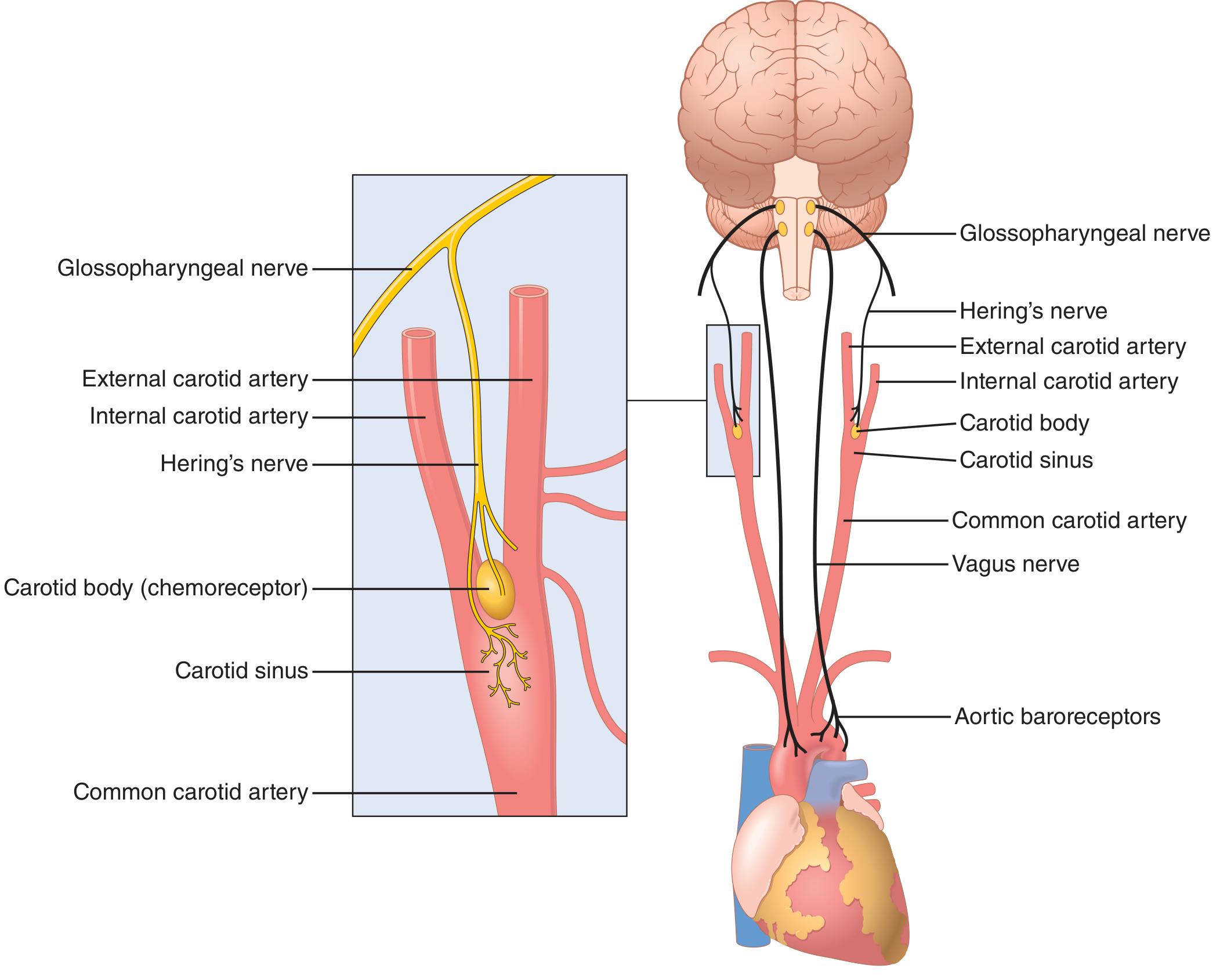
Guyton Fig. 18.5 - Anatomy of baroreceptor reflex arc
Sympathetic activation produces:
- Tachycardia and increased contractility
- Peripheral and renal vasoconstriction
- Renin release → Angiotensin II → Aldosterone
- Stimulation of ADH release from the posterior pituitary
This is the body's attempt to restore cardiac output (shown in the classic Guyton cardiac output curve):
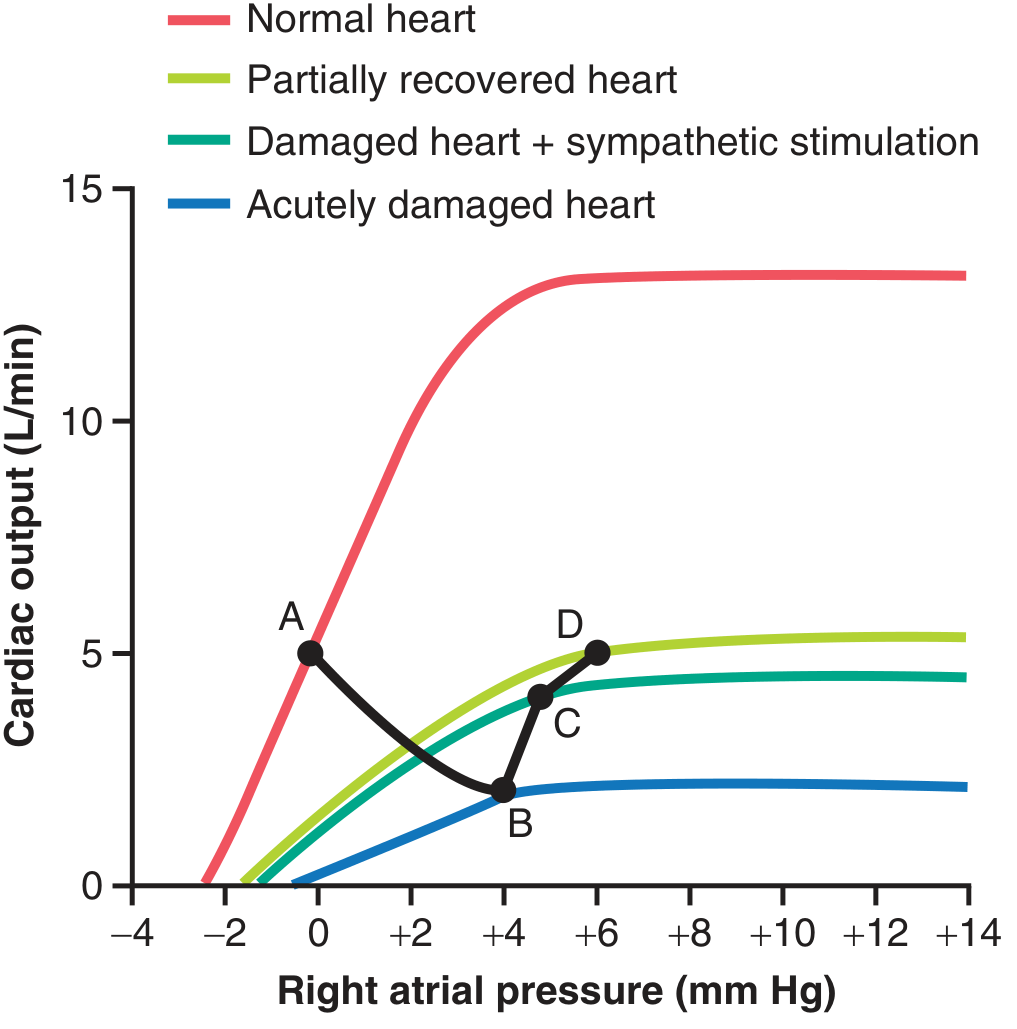
Guyton Fig. 22.1 - Progressive changes after acute MI. Point B→C: baroreceptor reflex rescues cardiac output. Point D: compensated HF with elevated right atrial pressure.
2. The Five Renal Mechanisms Retaining Fluid (Chronic HF)
Guyton lists five mechanisms by which the kidneys retain salt and water in HF (Ch. 22, pp. 282-283):
| # | Mechanism | Key Details |
|---|---|---|
| 1 | Reduced GFR | Low cardiac output → low afferent arteriolar pressure → decreased glomerular filtration; CO at half-normal can cause near-anuria |
| 2 | RAAS activation | Low renal blood flow → renin → Ang II → direct tubular Na/water reabsorption + peritubular capillary pressure drop |
| 3 | Aldosterone | Ang II stimulates adrenal cortex; hyperkalemia (from reduced GFR) also stimulates aldosterone; Na reabsorption in collecting tubule |
| 4 | ADH (key for osmoreceptors) | See below |
| 5 | Sympathetic activation | Renal arteriolar constriction, tubular Na reabsorption via α-receptors, renin stimulation, ADH stimulation |
3. ADH and Osmoreceptors in HF - The Non-Osmotic Override
Guyton & Hall (Ch. 22, p. 282) state directly:
"ADH is secreted by the hypothalamic-posterior pituitary gland system in response to increased extracellular fluid osmolarity, as well as nonosmotic stimuli from low-pressure (e.g., left atrial) and high-pressure (e.g., carotid sinus) baroreceptors. In severe heart failure, the nonosmotic effects of reductions in cardiac output and arterial pressure may predominate to stimulate secretion of ADH, which in turn causes excess water retention and hyponatremia (low plasma sodium concentration). Inappropriately high levels of ADH and hyponatremia are predictors for worsening outcomes in patients with heart failure."
Two control systems for ADH, compared:
| Control System | Normal Role | In HF |
|---|---|---|
| Osmoreceptors (hypothalamic) | Rise in plasma osmolality → ADH release → water retention to dilute ECF | Overridden; plasma osmolality may be LOW (dilutional) but ADH is still high |
| Baroreceptors (carotid sinus + atrial low-pressure receptors) | Pressure drop → non-osmotic ADH stimulus | Dominant driver in HF; reduced stretch → sustained non-osmotic ADH secretion |
From the Body Fluid chapter (Ch. 30), Guyton explains ADH's action: it allows the kidneys to reabsorb free water at the collecting duct (via V2 receptors), forming concentrated urine and expanding ECF volume. In HF, this free-water retention dilutes serum sodium, explaining dilutional hyponatremia.
Why osmoreceptors are "overridden": Osmoreceptors are osmolality sensors in the hypothalamus - they suppress ADH when osmolality is low. But the baroreceptor-driven (volume/pressure) signal to the pituitary is a stronger, parallel input that overrides this osmotic suppression. The result: ADH is secreted even when plasma is already hypoosmolar.
4. How Diuretics Correct These Disturbances
Guyton (Ch. 22) explains the treatment principle graphically and mechanistically:
Primary effect - reducing fluid overload and venous pressure:
"Reduction of pulmonary vascular congestion with diuretics may actually improve oxygenation and thereby improve myocardial function. Reduction of preload can reduce the size of the heart, allowing it to work at a more efficient fiber length." (Katzung, confirmed by Guyton's Frank-Starling logic)
In Guyton's cardiac output/venous return framework:
- Loop diuretics reduce blood volume → mean systemic filling pressure falls → venous return curve shifts left/downward
- This reduces right atrial pressure (preload), allowing the Frank-Starling curve to operate more efficiently
- A smaller, less overstretched heart pumps more effectively
Effect on Baroreceptors (indirect):
Effective diuresis improves forward cardiac output. As arterial pressure recovers toward normal:
- Carotid sinus and aortic arch baroreceptors are better stretched
- Firing rate increases → more inhibitory signals reach vasomotor center
- Sympathetic discharge is progressively withdrawn (pulse rate falls, cold clammy skin resolves)
- RAAS and renin activity decrease
Guyton notes this explicitly: "As the heart recovers, the fast pulse rate, cold skin, and pallor resulting from sympathetic stimulation in the acute stage of heart failure gradually disappear."
Effect on ADH/Osmoreceptor pathway:
By improving cardiac output and arterial pressure, diuretics reduce the non-osmotic baroreceptor drive for ADH release. With less ADH:
- Free water is no longer excessively retained at the collecting duct
- Plasma osmolality tends to normalize
- Once osmolality is normal, the hypothalamic osmoreceptors appropriately suppress further ADH secretion
- Dilutional hyponatremia improves
Guyton's body fluid chapter (Ch. 30) underlines this: when baroreceptor unloading is the cause of ADH excess, any intervention that restores effective arterial blood volume (diuretics, by reducing the total burden on the heart) also restores normal osmoreceptor-baroreceptor balance for ADH control.
The Danger - Excessive Diuresis:
Guyton's graphic analysis (venous return curves) teaches the critical limit: cardiac output in compensated HF is maintained by elevated filling pressures. If diuretics reduce filling pressure too aggressively:
- The heart slides to a lower point on its (already depressed) Frank-Starling curve
- Cardiac output falls
- Baroreceptors are once again unloaded → sympathetic reactivation, ADH resurgence
- Effective perfusion of kidneys falls → worsened renal function
Summary: Guyton & Hall Framework in One Flow
↓ Cardiac Output (HF)
│
▼
Baroreceptors unloaded (carotid sinus + aortic arch)
│
├─→ ↑ Sympathetic → tachycardia, vasoconstriction, ↑ renin → RAAS → aldosterone
│
└─→ Non-osmotic ADH release (overrides osmoreceptors)
│
└─→ Free water retention → dilutional hyponatremia
│
└─→ ↑ Venous pressure → edema/congestion
│
┌─────────────────┴──────────────────┐
│ │
DIURETICS act here (over-diuresis worsens)
│
▼
↓ Blood volume → ↓ venous return
│
▼
↓ Preload → heart works at better fiber length → ↑ CO
│
▼
Baroreceptors reloaded → ↓ sympathetic → ↓ non-osmotic ADH
│
▼
Osmoreceptors resume normal ADH control
│
▼
Congestion relieved; Na normalizes
Source: Guyton and Hall Textbook of Medical Physiology - Chapter 22 (Heart Failure, pp. 277-283) and Chapter 30 (Body Fluid Regulation: Baroreceptor and Sympathetic Reflex Control of Renal Excretion, pp. 401-402; Role of ADH, pp. 403-404)
This is a shared conversation. Sign in to Orris to start your own chat.