Neuromuscular junction and myasthenia gravis
Reading File
Finding Sources
Finding Sources
Searching PubMed
"myasthenia gravis"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Reading File
Loading Image
Reading File
I now have comprehensive information. Let me compile the full answer:
Neuromuscular Junction & Myasthenia Gravis
PART 1 - THE NEUROMUSCULAR JUNCTION
Structure
The neuromuscular junction (NMJ) is the specialized synapse where a motor nerve terminates on a skeletal muscle fiber. As the motor axon approaches its target, it loses its myelin sheath and branches into multiple terminal boutons. Each terminal bouton:
- Contains thousands of synaptic vesicles loaded with acetylcholine (ACh)
- Sits within junctional folds (depressions in the motor endplate)
- Is separated from the postsynaptic muscle membrane by the synaptic cleft
The postsynaptic muscle membrane (sarcolemma) is heavily folded, and nicotinic ACh receptors (N_M) are concentrated at the tops of these junctional folds. Acetylcholinesterase (AChE) is anchored at the base of the folds and in the cleft.
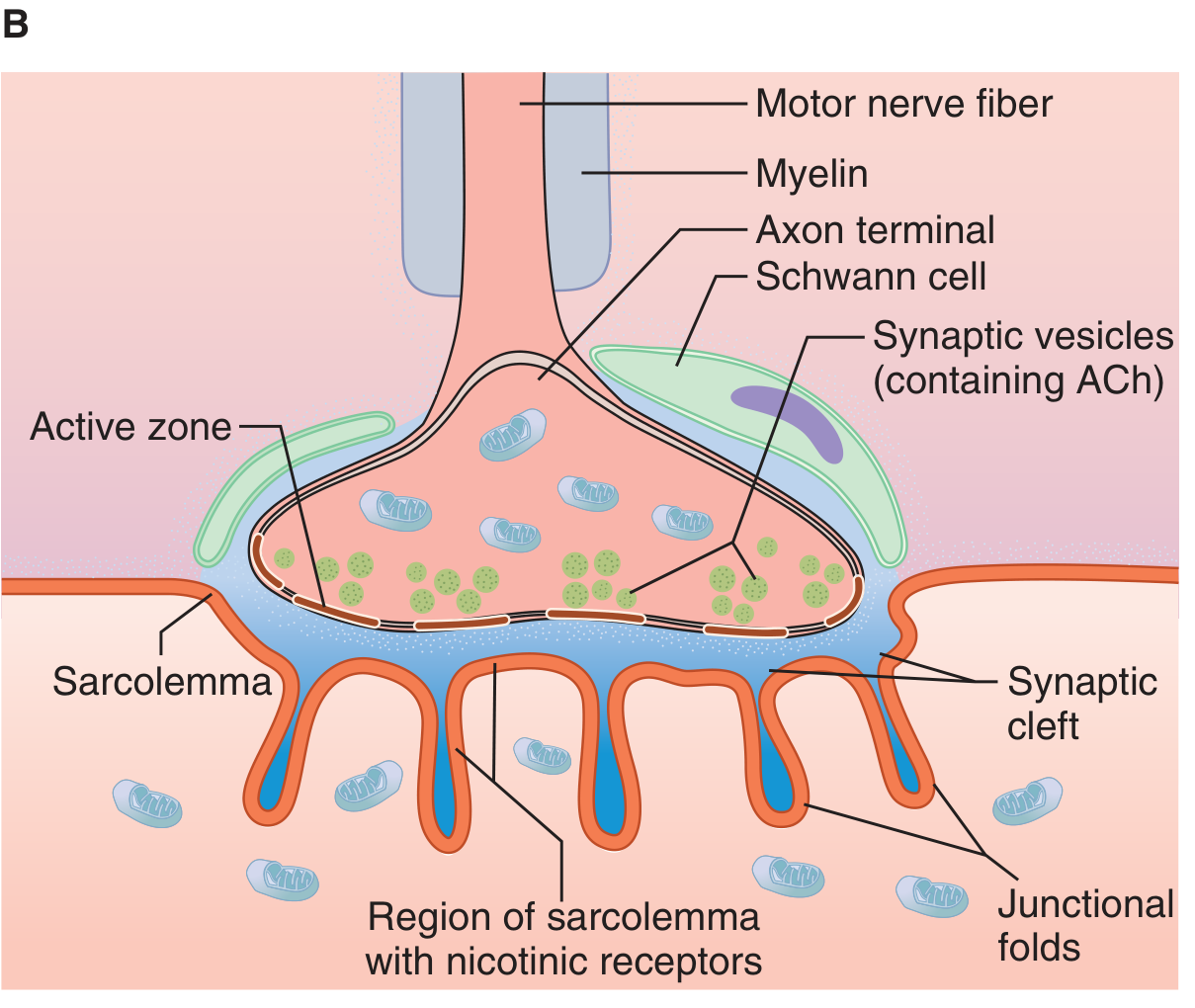
NMJ structure: motor nerve fiber, axon terminal, synaptic vesicles (ACh), synaptic cleft, junctional folds, and nicotinic receptors on the sarcolemma. (Ganong's Review of Medical Physiology, 26e)
Transmission Events (Step by Step)
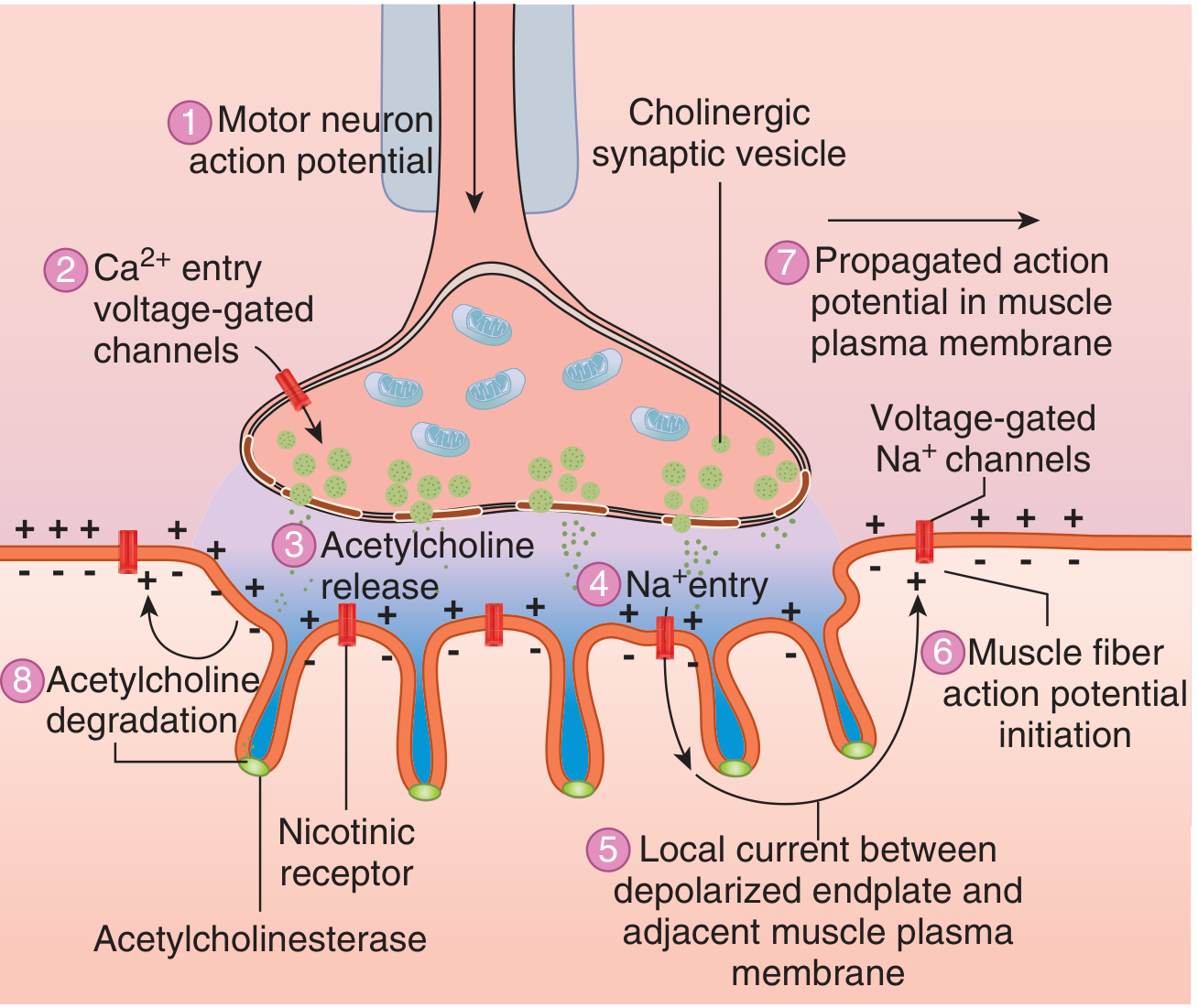
8 sequential events at the NMJ leading to muscle action potential. (Ganong's Review of Medical Physiology, 26e)
| Step | Event |
|---|---|
| 1 | Motor neuron action potential arrives at axon terminal |
| 2 | Voltage-gated Ca²+ channels open; Ca²+ floods into the terminal |
| 3 | Ca²+ triggers exocytosis of ACh-containing synaptic vesicles |
| 4 | ACh diffuses across the synaptic cleft and binds to postsynaptic nicotinic (N_M) receptors |
| 5 | Receptor binding increases Na⁺ and K⁺ conductance; net Na⁺ influx produces the endplate potential (EPP) |
| 6 | The current sink created by the EPP depolarizes adjacent muscle membrane to its firing threshold |
| 7 | Voltage-gated Na⁺ channels trigger a propagated action potential along the sarcolemma |
| 8 | ACh in the cleft is rapidly hydrolyzed by acetylcholinesterase (into choline + acetate); choline is recycled back into the nerve terminal |
- Ganong's Review of Medical Physiology, 26e
- Goodman & Gilman's Pharmacological Basis of Therapeutics
The Nicotinic AChR (N_M)
The muscle-type nicotinic receptor is a pentameric ligand-gated ion channel composed of α₁, α₁, β, δ, and ε (adult) or γ (fetal) subunits. Two ACh molecules must bind (one to each α subunit) to open the channel. This allows Na⁺ influx and K⁺ efflux, generating the EPP. The main immunogenic region (MIR) on the α subunit is the primary target of autoantibodies in MG.
PART 2 - MYASTHENIA GRAVIS
Definition
Acquired myasthenia gravis (MG) is the most common primary disorder of neuromuscular transmission. Autoantibodies - most commonly against the AChR - disrupt normal NMT, resulting in fatigable muscle weakness that worsens with activity and improves with rest. - Bradley and Daroff's Neurology in Clinical Practice
Epidemiology
- Prevalence ~20/100,000 (~60,000 US patients)
- Bimodal age distribution:
- Peak 1: Young women (< 40 years) - associated with thymic hyperplasia
- Peak 2: Older men (> 50 years) - may have thymoma
- Women are affected ~3x more than men before age 40; incidence is roughly equal after age 50
- Overall prevalence is rising (better ascertainment, improved survival, aging population)
- Bradley and Daroff's Neurology in Clinical Practice | Medical Physiology (Boron & Boulpaep)
Pathophysiology
Anti-AChR antibodies (present in ~80-85% of patients) act by three mechanisms:
- Complement-mediated destruction of junctional folds - loss of postsynaptic architecture
- Accelerated internalization and degradation of AChR
- Blocking of ACh-AChR binding (a smaller subset)
The net result is a reduced surface density of functional AChR, smaller endplate potentials, and failure to reach threshold at a significant fraction of NMJs during sustained activity - producing the characteristic fatigable weakness.
The amplitude of miniature endplate potentials (MEPPs) is reduced in MG, though quantal frequency is normal - pointing clearly to a postsynaptic defect. - Medical Physiology (Boron & Boulpaep)
The thymus plays a central role: thymic cells express AChR subunits (especially the α subunit), and aberrant thymic T cell activation initiates or perpetuates the autoimmune response.
Clinical Subtypes and Antibodies
| MG Subtype | Age at Onset | Thymic Histology | Autoantibodies | Notes |
|---|---|---|---|---|
| Ocular | Adult (US/EU); childhood (Asia) | Unknown | AChR (50%) | Weakness restricted to ocular muscles |
| Early Onset | < 50 years | Hyperplasia | AChR | Female > Male (1:3) |
| Late Onset | > 50 years | Normal | AChR, Titin | Male > Female |
| Thymoma | > 40 years | Neoplasia | AChR, Titin, Ryanodine | Anti-titin = severe disease |
| MuSK | < 40 years | Normal | MuSK | Oropharyngeal/facial/respiratory predominance; ChEI may worsen |
| LRP4 | 30-50 years | Unknown | LRP4 | Marked female predominance; IgG4 antibodies |
| Seronegative | Variable | Hyperplasia in some | Low-affinity AChR Abs, Agrin, Cortactin in some | Managed similarly to AChR-MG |
Bradley and Daroff's Neurology in Clinical Practice, Table 108.1
Clinical Presentation
Cardinal feature: Fatigable muscle weakness - worst at end of day or after exertion; improved by rest.
Initial symptoms:
- ~2/3: Ptosis or diplopia (ocular involvement) - the most common presentation
- ~1/6: Dysphagia, dysarthria, or jaw weakness
- ~10%: Limb weakness
- ~10-15%: Remain purely ocular (higher rate in Asian populations/children, up to 58%)
Key ocular signs:
- Ptosis (often asymmetric, shifts from eye to eye - virtually pathognomonic of MG)
- Ophthalmoplegia without pupillary involvement
- "Lid twitch" (Cogan's sign): after downgaze, brief lid overshoot on upgaze
- No pupillary abnormality (distinguishes from CN III palsy)

Ocular motility abnormalities in MG: ptosis, ophthalmoplegia, and asymmetric eye movements in various gaze directions. (Bradley and Daroff's Neurology in Clinical Practice)
Other affected muscle groups:
- Bulbar: jaw, facial, palatal, pharyngeal (nasal voice, regurgitation)
- Neck flexors (weaker than extensors)
- Proximal limbs: deltoids, hip flexors
- Respiratory muscles (myasthenic crisis)
Aggravating factors: Systemic illness, emotional stress, heat, thyroid disease, pregnancy, menstrual cycle, certain drugs (aminoglycosides, fluoroquinolones, beta-blockers, Mg²+, curare-type agents)
Diagnosis
1. Antibody testing:
- Anti-AChR binding antibodies: Positive in ~80-85% of generalized MG; ~50% of ocular MG. Virtually confirms the diagnosis when positive (false positives are rare but occur in lupus, autoimmune liver disease, ALS, thymoma without MG).
- Anti-MuSK antibodies: Present in ~50% of AChR-seronegative generalized MG
- Anti-LRP4 antibodies: A subset of double-seronegative patients
- Anti-titin/ryanodine antibodies: Predict thymoma; associated with severe disease
- AChR modulating antibodies: Detect ~10% of AChR binding Ab-negative patients
2. Edrophonium (Tensilon) test: IV edrophonium (short-acting AChEI) produces transient improvement in weakness (especially ptosis). Largely replaced by antibody testing due to false positives/negatives and cardiac risks.
3. Electrodiagnostic testing:
- Repetitive nerve stimulation (RNS): Characteristic decremental response (>10% decrement at 2-3 Hz)
- Single-fiber EMG (SFEMG): Most sensitive test - detects increased "jitter" (variability in NMJ transmission); abnormal in >95% of generalized MG
4. Chest CT/MRI: Mandatory in all MG patients to look for thymoma (found in ~10-15% of MG patients)
Treatment
Treatment is organized into four categories:
A. Symptomatic - Cholinesterase Inhibitors (ChEIs)
- Pyridostigmine (Mestinon): First-line symptomatic therapy; inhibits AChE, prolongs ACh action in the cleft. Dose: 30-60 mg every 4-6 hours. Overdose causes cholinergic crisis (muscarinic: excess secretions, bradycardia, diarrhea; nicotinic: muscle fasciculations, weakness).
- Caution in MuSK-MG: ChEIs may worsen symptoms, with profuse fasciculations.
B. Rapid, Short-Term Immunotherapy (Rescue)
- Plasmapheresis (PLEX): Removes circulating antibodies; rapid onset (days), lasts weeks. Used in myasthenic crisis or pre-operatively.
- IV immunoglobulin (IVIg): Comparable efficacy to PLEX; exact mechanism unclear (Fc receptor blockade, complement inhibition, anti-idiotype antibodies).
C. Long-Term Immunosuppression
- Corticosteroids (Prednisone): Most widely used. Initial transient worsening is common before improvement. Typically started low and escalated.
- Azathioprine: Steroid-sparing agent; onset 6-12 months. Monitor CBC and LFTs.
- Mycophenolate mofetil: Alternative steroid-sparing agent.
- Cyclosporine: Used in refractory cases.
- Rituximab (RTX): Particularly effective in MuSK-MG; recent Cochrane review (2025, PMID: 40607605) supports its use.
D. Novel Targeted Therapies
- FcRn inhibitors (efgartigimod, rozanolixizumab, nipocalimab): Reduce IgG (and hence autoantibody) levels by blocking neonatal Fc receptor-mediated IgG recycling. 2025 meta-analysis (PMID: 40288289) confirms efficacy and safety.
- Complement inhibitors (eculizumab, ravulizumab): Block the complement cascade that destroys junctional folds in AChR-ab MG.
E. Thymectomy
- Indicated for all thymoma patients (removes source of antigenic stimulation and/or neoplasm).
- Benefits patients with AChR-positive generalized MG, especially those with thymic hyperplasia (early-onset).
- Thymectomy leads to clinical improvement in ~75% of patients with thymoma. The MGTX randomized trial confirmed benefit in non-thymomatous AChR-positive generalized MG.
- Role in MuSK-MG is not yet established.
Myasthenic Crisis vs. Cholinergic Crisis
| Feature | Myasthenic Crisis | Cholinergic Crisis |
|---|---|---|
| Cause | Inadequate treatment / trigger | Excess ChEI |
| Muscle weakness | Yes | Yes |
| Pupils | Normal/dilated | Miotic |
| Secretions | Normal | Increased (SLUDGE) |
| Heart rate | Normal/fast | Bradycardia |
| Response to edrophonium | Improves weakness | Worsens weakness |
| Treatment | Intubation + PLEX/IVIg | Stop ChEI; atropine |
Differential Diagnosis
- Lambert-Eaton Myasthenic Syndrome (LEMS): Presynaptic disorder (anti-VGCC antibodies); proximal limb weakness, depressed reflexes that augment with exercise; often paraneoplastic (small cell lung cancer)
- Ocular MG vs. internuclear ophthalmoplegia (MS) - no fatigability in INO
- Botulism: Presynaptic; pupil involvement, descending paralysis
- Congenital myasthenic syndromes: No autoantibodies; present at birth or early childhood
- Miller Fisher syndrome (GBS variant): Ophthalmoplegia + ataxia + areflexia; anti-GQ1b antibodies
Key Points Summary
- The NMJ converts an electrical nerve signal into muscle contraction via Ca²+-triggered ACh release → EPP → muscle action potential
- MG is an autoimmune disease targeting postsynaptic AChR (80-85%), MuSK, or LRP4
- Cardinal feature: fatigable, fluctuating weakness - worse with activity, better with rest
- Hallmark diagnostic features: positive AChR-ab, decremental RNS response, increased SFEMG jitter
- First-line symptomatic treatment: pyridostigmine
- Long-term: corticosteroids ± steroid-sparing immunosuppressants
- Always image the chest for thymoma
- New biologic agents (FcRn inhibitors, complement inhibitors, rituximab) are transforming management of refractory MG
Sources: Ganong's Review of Medical Physiology, 26e | Bradley and Daroff's Neurology in Clinical Practice | Medical Physiology (Boron & Boulpaep) | Goodman & Gilman's Pharmacological Basis of Therapeutics | Recent PubMed: FcRn inhibitor meta-analysis PMID 40288289, Rituximab in MG Cochrane review PMID 40607605
This is a shared conversation. Sign in to Orris to start your own chat.