Coronary circulation and oxygen demand imbalance under anesthesia 20 marks answer
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
I now have comprehensive material from multiple authoritative textbooks. Let me compose the full 20-mark answer.
Coronary Circulation and Oxygen Demand-Supply Imbalance Under Anesthesia
1. ANATOMY OF THE CORONARY CIRCULATION
The coronary arteries arise from the sinuses of Valsalva at the root of the aorta and lie on the epicardial surface, sending intramuscular branches into the myocardium.
- Left coronary artery (LCA): Divides into the left anterior descending (LAD) and circumflex (Cx) arteries. The LAD supplies the anterior wall and septum of the left ventricle; the Cx supplies the lateral and posterior LV wall.
- Right coronary artery (RCA): Traverses the right AV groove, supplying the right ventricle via acute marginal branches. In 80-90% of people ("right dominant"), the RCA gives rise to the posterior descending artery (PDA) that supplies the inferior LV. The AV node artery arises from the dominant vessel; the SA node artery arises from the RCA in 55% of individuals.
- Venous drainage: ~75% of left ventricular coronary venous blood returns via the coronary sinus to the right atrium. Right ventricular blood returns largely via anterior cardiac veins directly to the right atrium. A small amount drains through thebesian veins directly into all cardiac chambers.
- Resting coronary blood flow averages ~70 mL/min/100 g (~225 mL/min total), representing 4-5% of cardiac output.
(Guyton & Hall Medical Physiology; Barash's Clinical Anesthesia 9e)
2. PHASIC NATURE OF CORONARY BLOOD FLOW
A defining feature of the coronary circulation is the biphasic pattern determined by the cardiac cycle:
| Phase | LV Coronary Flow | Mechanism |
|---|---|---|
| Systole | Markedly reduced | Extravascular compression of intramural vessels by LV contraction raises vascular resistance |
| Diastole | Maximal (70-80% of total flow) | Myocardium relaxes; resistance falls; flow surges |
The right ventricle, generating much lower pressures, does not suffer the same systolic impedance - its flow pattern resembles that of peripheral vascular beds.
The coronary perfusion pressure (CPP) for the LV = Aortic diastolic pressure - LV end-diastolic pressure (LVEDP). This has immediate clinical relevance: both hypotension (lowering diastolic Ao P) and elevated LVEDP (from heart failure, volume overload, or ischemia itself) reduce CPP and worsen perfusion.
The subendocardium is most vulnerable because:
- It is furthest from epicardial supply vessels
- Intramural pressure is highest in the subendocardium
- Its flow reserve is recruited first and exhausted soonest under stress
(Barash's Clinical Anesthesia 9e, p.857; Guyton & Hall)
3. DETERMINANTS OF MYOCARDIAL OXYGEN SUPPLY
Myocardial oxygen supply depends on two factors: coronary blood flow and arterial oxygen content.
3a. Coronary Blood Flow
Blood flow = Coronary Perfusion Pressure / Coronary Vascular Resistance
Resistance is modulated by:
- Metabolic (local) autoregulation - the dominant controller. Hypoxia and adenosine cause coronary arteriolar vasodilation. Carbon dioxide (from Krebs cycle), reactive oxygen species, and nitric oxide (NO) from eNOS are upstream mediators. K_ATP channels (responding to falling ATP) cause smooth muscle hyperpolarization and vasodilation.
- Autoregulation of flow: Coronary blood flow is maintained constant over a wide range of perfusion pressures (approximately 60-130 mmHg). Coronary flow reserve (CFR) is the ratio of maximum achievable flow to resting flow - normally 4-6x. As stenosis worsens, CFR narrows.
- Endothelium-derived factors: Prostacyclin (PGI2) causes vasodilation and inhibits platelet aggregation. Endothelin-1 causes vasoconstriction but is normally counterbalanced by NO. Atherosclerosis impairs NO release, reducing vasodilatory capacity.
- Neural control: Sympathetic stimulation (alpha-1) causes mild direct vasoconstriction, but beta-2 stimulation and the dominant metabolic vasodilation generally prevail. Net effect during stress is vasodilation driven by increased metabolic demand.
3b. Arterial Oxygen Content
CaO2 = (Hb × 1.34 × SaO2) + (0.003 × PaO2)
- Hemoglobin concentration is the primary determinant
- The oxyhemoglobin dissociation curve position matters: leftward shift (alkalosis, hypothermia, low 2,3-DPG) decreases oxygen release to tissues even at adequate delivery
- Dissolved oxygen contributes minimally at normal PaO2
(Miller's Anesthesia 10e; Barash's Clinical Anesthesia 9e)
4. DETERMINANTS OF MYOCARDIAL OXYGEN DEMAND
The heart is an obligate aerobic organ. It derives ~75% of oxygen from hemoglobin under basal conditions (coronary sinus PO2 ≈ 20 mmHg) - far exceeding other organs. Because near-maximal oxygen extraction already occurs at rest, the only way to meet increased demand is by increasing coronary blood flow.
Major determinants of myocardial oxygen demand (MVO2):
| Determinant | Clinical Significance |
|---|---|
| Heart rate | Most important single factor - doubles diastolic filling time effect; tachycardia both increases demand AND reduces supply time |
| Wall tension (Laplace) | T = (Pressure × Radius) / (2 × Wall thickness); increases with afterload (hypertension) or preload (dilation) |
| Contractility (inotropy) | Catecholamine surge, light anesthesia raise MVO2 |
| Basal metabolic rate | Relatively fixed component (ion pump maintenance) |
Contraction will cease within 10-15 seconds of acute coronary occlusion unless sufficient collateral flow exists. This tight coupling of flow to metabolism explains why any anesthetic perturbation disrupting the supply-demand balance is potentially dangerous.
(Barash's Clinical Anesthesia 9e, p.859)
5. HOW ANESTHESIA DISRUPTS THE OXYGEN BALANCE
5a. Hemodynamic Effects
Hypotension (the commonest anesthetic cause of ischemia):
- Reduced aortic diastolic pressure decreases CPP
- Common causes: volatile anesthetic vasodilation, neuraxial sympathectomy, hypovolemia, myocardial depression, aortocaval compression
Tachycardia:
- A dual threat: increases oxygen demand AND reduces diastolic time, shrinking the window for coronary perfusion
- Even small heart rate increases during the perioperative period are clinically significant, especially with underlying CAD
- This is the physiological basis for beta-blocker use as anti-ischemic therapy
Hypertension (from light anesthesia or surgical stimulation):
- Raises afterload and wall tension, sharply increasing MVO2
- Paradoxically, it may increase CPP but the demand side usually predominates
Elevated LVEDP:
- From myocardial depression, volume loading, or ischemia itself
- Directly compresses subendocardial vessels, worsening the most vulnerable zone
5b. Respiratory and Hematological Factors
- Hypoxia - reduces arterial oxygen content; also a direct coronary vasodilator (compensatory) but fails when flow is limited by stenosis
- Anemia - reduced oxygen-carrying capacity; critical in patients with limited coronary reserve
- Alkalosis / Hypothermia - leftward shift of oxygen-hemoglobin dissociation curve, impairs O2 delivery to myocardium even when hemoglobin is saturated
- Hypercapnia - coronary vasodilator but also causes sympathetic activation (raises demand)
5c. Sympathetic Nervous System Activation
Surgical stress, laryngoscopy, intubation, and inadequate analgesia activate the sympathetic axis. Catecholamine surge causes:
- Coronary vasoconstriction (alpha-1 on large arteries)
- Tachycardia and hypertension (increased demand)
- In diseased vessels with impaired endothelium, vasoconstriction can be paradoxically exaggerated
(Sabiston Textbook of Surgery; Miller's Anesthesia 10e)
6. VOLATILE ANESTHETIC AGENTS AND CORONARY CIRCULATION
Coronary Steal (Isoflurane Controversy)
Isoflurane is a potent coronary vasodilator. Theoretical concern was "coronary steal": vasodilation of vessels supplying well-perfused territories, diverting blood away from ischemic zones (already maximally vasodilated, distal to a stenosis) to normal zones.
Definition: Steal = diversion of blood from a poorly perfused bed (with limited or absent autoregulation) to a better-perfused bed retaining autoregulatory capacity.
Clinical reality: Large outcome studies in patients undergoing CABG have found no significant increase in myocardial infarction or perioperative death with isoflurane. In a chronically instrumented dog model with multivessel obstruction, neither isoflurane, sevoflurane, nor desflurane at up to 1.5 MAC caused abnormal collateral flow redistribution, whereas adenosine (a pure vasodilator) clearly did.
- Sevoflurane is approximately half as potent a coronary vasodilator as isoflurane
- Desflurane - when given without opioids at high concentrations in CAD patients, has caused significant ischemia, necessitating beta-blockers. Rapid increases in inspired desflurane concentration trigger sympathetic activation with tachycardia and hypertension - both unfavorable for the supply-demand balance
- Determinants of myocardial oxygen supply and demand, rather than the choice of volatile agent, are of greater importance to patient outcomes
(Barash's Clinical Anesthesia 9e, p.1427-1428)
Anesthetic Preconditioning (Cardioprotection)
Volatile anesthetics given before (preconditioning) or immediately after (postconditioning) ischemia mimic ischemic preconditioning:
- They diffuse through myocardial cell membranes and alter mitochondrial electron transport, generating reactive oxygen species as triggers
- This activates Protein Kinase C → K_ATP channel opening
- ~30-40% of cardioprotection relates to reduced mitochondrial calcium loading
- Opioids (especially at high doses) maintain the O2 supply-demand ratio in cardiac surgery as well as or better than inhalation-based techniques; they also exhibit delta- and kappa-opioid receptor mediated preconditioning
7. PERIOPERATIVE MYOCARDIAL ISCHEMIA (PMI)
PMI complicates noncardiac surgery through two mechanisms:
- Plaque rupture and thrombosis - acute coronary syndrome pattern, often presenting as STEMI
- Demand-supply imbalance - more common perioperatively; typically subendocardial, NSTEMI pattern
Crucially, 65% of patients with PMI have no ischemic symptoms (POISE trial) - the anesthetic and pain medications mask pain, and patients are sedated or intubated. Silent ischemia is the rule, not the exception.
Timing: Most PMI occurs within 48 hours of surgery.
Precipitants under anesthesia:
- Hypotension, hypertension, tachycardia
- Bleeding and anemia
- Hypoxia and hypothermia
- Coronary vasoconstriction from catecholamines
(Sabiston Textbook of Surgery)
8. MONITORING FOR ISCHEMIA UNDER ANESTHESIA
ECG Monitoring
- Multilead ST-segment analysis - leads II and V5 together detect >80% of ischemic events; all five leads detect >95%
- ST depression ≥1 mm (subendocardial ischemia - demand-type)
- ST elevation (transmural ischemia / spasm)
- T-wave inversion, new LBBB, arrhythmias
Transesophageal Echocardiography (TEE)
- Regional wall motion abnormalities (RWMA) are the earliest sign of ischemia - appearing before ECG changes
- Segmental hypokinesia, akinesia, or dyskinesia in a coronary territory indicates ischemia
- Indispensable in cardiac surgery and high-risk noncardiac surgery
(Miller's Anesthesia 10e)
9. MANAGEMENT PRINCIPLES
| Goal | Intervention |
|---|---|
| Reduce heart rate | Beta-blockers (metoprolol, esmolol); continue perioperative beta-blockade |
| Maintain diastolic BP | Vasopressors (phenylephrine - pure alpha for reflex bradycardia; norepinephrine for combined) |
| Reduce LVEDP | Diuretics, nitroglycerin (venodilation reduces preload) |
| Reduce wall tension | Nitroglycerin, nitroprusside for afterload reduction |
| Increase O2 supply | Supplemental oxygen, correct anemia (transfusion threshold debated; Hb ~8-10 in CAD) |
| Avoid tachycardia | Adequate analgesia, avoid light anesthesia, avoid rapid desflurane escalation |
| Coronary vasodilation | IV nitroglycerin (also relieves coronary spasm) |
| Cardiac support if failing | IABP (augments diastolic CPP; reduces systolic afterload) |
Revised Cardiac Risk Index (RCRI) - 6 independent predictors of PMI:
- High-risk surgery (intraperitoneal, intrathoracic, suprainguinal vascular)
- History of ischemic heart disease
- History of congestive heart failure
- History of cerebrovascular disease
- Insulin-dependent diabetes
- Serum creatinine >2.0 mg/dL
Patients with ≥2 risk factors require more intensive perioperative monitoring and optimization.
SUMMARY DIAGRAM (Conceptual)
CORONARY OXYGEN BALANCE UNDER ANESTHESIA
│
┌───────────┴───────────┐
O2 SUPPLY O2 DEMAND
│ │
┌─────┴──────┐ ┌──────┴──────┐
CBF × CaO2 HR × WallTension × Contractility
│ │ │ │
CPP= Hb, SaO2 Anesthetic Sympathetic
AoD- Leftward shift depth activation
LVEDP → impairs O2 Opioids (laryngoscopy,
release reduce light plane)
│ demand
Tachycardia = ↓diastole
Hypotension = ↓AoD
↑LVEDP = ↓CPP
→ SUBENDOCARDIAL ISCHEMIA
CITATIONS
- Miller's Anesthesia, 10th Edition - Determinants of Myocardial Oxygen Supply and Demand; Coronary Circulation and Myocardial Ischemia
- Barash's Clinical Anesthesia, 9th Edition - Coronary Physiology; Coronary Steal, Myocardial Ischemia, and Cardiac Outcome; Cardioprotection from Volatile Anesthetics
- Guyton & Hall Textbook of Medical Physiology - Coronary Circulation, Control of Coronary Blood Flow
- Costanzo Physiology, 7th Edition - Coronary Circulation
- Sabiston Textbook of Surgery - Perioperative Myocardial Ischemia and Infarction
Explain concept of context sensitivity half time. How does this concept apply different in propofol and fentanyl in clinical practice of anesthesia. 20 marks answer
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Finding Sources
Finding Sources
Reading File
I now have comprehensive material from four authoritative anesthesia textbooks plus the key CSHT graph. I can now compose the full 20-mark answer.
Context-Sensitive Half-Time: Concept, Propofol vs Fentanyl in Clinical Anesthesia
1. THE PROBLEM WITH ELIMINATION HALF-LIFE (t½)
The classical pharmacokinetic parameter, elimination half-life (t½), describes the time for plasma drug concentration to fall by 50% - but it was derived from a one-compartment model and applies strictly only to a single-dose situation. In clinical anesthesia, drugs are almost always given by continuous infusion to maintain effect-site concentrations throughout a procedure. For multi-compartment drugs (which most anesthetics are), the elimination t½ fails completely to predict when a patient will wake up after an infusion is stopped:
- It does not account for drug redistribution from peripheral tissue compartments back into the plasma after the infusion ends
- It does not account for how long the infusion has been running (saturation of peripheral compartments)
- It gives a fixed number that is infusion-duration independent, which is misleading
A new concept was needed - and was provided by Hughes, Glass, and Jacobs in 1992.
(Barash's Clinical Anesthesia 9e; Barash's Clinical Anesthesia 9e, p.1470)
2. DEFINITION OF CONTEXT-SENSITIVE HALF-TIME (CSHT)
Context-Sensitive Half-Time is defined as the time required for the plasma drug concentration to decline by 50% after the termination of a continuous infusion of a given duration.
The word "context" refers to the duration of the infusion - the context under which the drug was administered. Different infusion durations create different tissue saturation states, so the CSHT varies with context (infusion duration). This parameter is:
- Calculated by computer simulation of multicompartmental pharmacokinetic models
- A function of both distribution (drug returning from peripheral compartments) and elimination (hepatic/other clearance)
- Expressed as a curve plotting CSHT (y-axis, minutes) against infusion duration (x-axis, hours) - not a single number
(Barash's Clinical Anesthesia 9e, p.2509)
3. MULTICOMPARTMENT PHARMACOKINETIC FRAMEWORK
To understand CSHT, the three-compartment model must be understood:
DOSE → [Central Compartment (V1)] ← → [Rapid Peripheral (V2)]
Plasma / Blood Muscle, viscera
↕
[Slow Peripheral (V3)]
Fat, bone
↕
Elimination (clearance)
During infusion:
- Drug distributes from central compartment → peripheral compartments (V2, V3)
- Plasma concentration is buffered by this outward distribution
After infusion stops:
- Drug flows back from peripheral compartments → central compartment (down the concentration gradient)
- This return flow replaces drug eliminated from plasma, slowing the fall in plasma concentration
- The longer the infusion, the more drug is stored in peripheral compartments, the more returns = the longer the CSHT
The critical insight: the CSHT captures this interplay between redistribution and elimination that a single t½ value cannot.
4. THE CLASSIC CSHT GRAPH (Hughes et al., 1992)
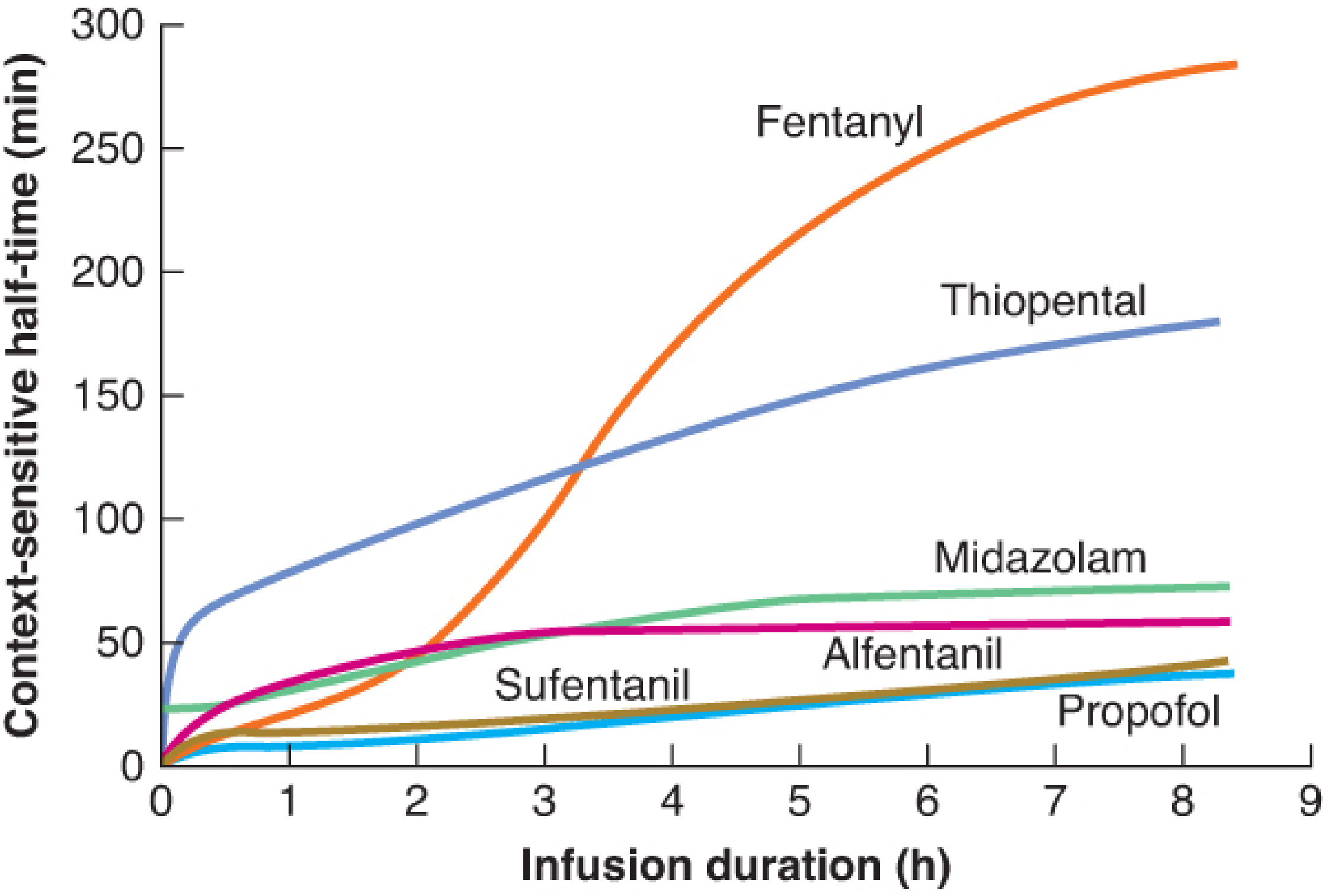
Figure: Context-sensitive half-time as a function of infusion duration. Note the dramatically divergent behaviors of fentanyl (rises steeply to >280 min at 8 h) versus propofol (remains flat near 30-40 min). - Barash's Clinical Anesthesia 9e, Figure 30-4
Key observations from this graph:
- All drugs show increasing CSHT with longer infusions - but to vastly different degrees
- Fentanyl shows the steepest and most dramatic rise - approaching 280+ minutes after an 8-hour infusion
- Propofol shows a nearly flat curve - remaining below ~40 minutes even after 8+ hours of infusion
- Thiopental rises substantially but more slowly than fentanyl
- Sufentanil stays far below fentanyl despite having a longer elimination t½ - this counterintuitive finding is a key teaching point
5. PROPOFOL: CLINICAL PHARMACOKINETICS AND CSHT
Why Propofol has a Favorable (Low) CSHT
Propofol's pharmacokinetic behavior is determined by two properties acting in combination:
a) High Hepatic Metabolic Clearance
- Propofol undergoes rapid, extensive hepatic metabolism (glucuronidation and hydroxylation)
- Its clearance (1.5-2.0 L/min) exceeds hepatic blood flow - indicating extrahepatic metabolism as well (lung, kidney, gut)
- Drug returning from peripheral compartments is rapidly cleared before it can accumulate in plasma
- Net effect: the return of drug from tissues does not significantly impede plasma concentration decay
b) Relatively Slow Return from Peripheral Compartments
- Although propofol is highly lipophilic, the rate at which it returns from deep peripheral compartments (fat) is slow
- Combined with rapid elimination, even the drug that does return is immediately removed
Clinical implication: After even an 8-hour propofol infusion, the CSHT is only ~30-40 minutes. This makes propofol uniquely suitable as a continuous infusion hypnotic.
(Katzung Basic & Clinical Pharmacology 16e; Barash's Clinical Anesthesia 9e, p.2509)
Propofol in TIVA (Total Intravenous Anesthesia)
- Propofol's short CSHT is the primary reason it is the preferred drug for TIVA and target-controlled infusion (TCI)
- Its CSHT profile allows confident prediction of awakening regardless of infusion duration
- Used for induction (1-2.5 mg/kg IV) followed by maintenance infusion (25-75 mcg/kg/min)
- Has additional advantages: antiemetic properties, smooth emergence, reduced postoperative nausea/vomiting (PONV)
Effect-Site Equilibration (ke0) of Propofol
- Propofol has a short t½ ke0 (rapid equilibration between plasma and brain/effect site)
- This means plasma concentration changes are quickly reflected in clinical effect - making it easily titratable
- This short ke0 also means rapid onset of consciousness loss after bolus induction
6. FENTANYL: CLINICAL PHARMACOKINETICS AND CSHT
Why Fentanyl has an Unfavorable (High) CSHT
Fentanyl's behavior is fundamentally different and clinically deceptive:
a) Extreme Lipophilicity and Large Volume of Distribution
- Fentanyl is highly lipophilic (octanol:water partition coefficient ~816)
- Volume of distribution at steady state (Vdss) is very large (~4-6 L/kg)
- Rapid initial redistribution into fat, muscle, and other peripheral tissues causes the short clinical duration after a single bolus dose - NOT elimination
- This is redistribution, not metabolism - the drug is still present in the body in large amounts
b) Rapid Peripheral Saturation with Prolonged Infusion
- As infusion continues, peripheral compartments (especially fat) become progressively saturated
- Once saturation is reached, redistribution can no longer act as a buffer
- When infusion stops, enormous amounts of stored fentanyl return from peripheral compartments to plasma
- Hepatic clearance (~1 L/min) is insufficient to keep pace with this return
- Result: plasma concentration falls extremely slowly
c) The Sufentanil Paradox
A critical teaching point: Sufentanil has a longer elimination t½ than fentanyl (577 vs 462 minutes), yet has a much shorter CSHT for infusions >2 hours.
Why? Sufentanil has such high clearance that even though its peripheral compartments are large, the drug returning from those compartments is metabolized faster. Fentanyl's lower clearance relative to its tissue binding capacity makes it unable to keep up with the redistributed drug load.
(Barash's Clinical Anesthesia 9e, p.2509; Miller's Anesthesia 10e, p.2799)
Clinical Implications of Fentanyl's CSHT
| Infusion Duration | Approx. CSHT of Fentanyl |
|---|---|
| 30 min | ~20-25 min |
| 1 hour | ~50-60 min |
| 2 hours | ~100 min |
| 4 hours | ~200 min |
| 8 hours | ~280 min |
After a 1-hour continuous infusion, fentanyl's CSHT is nearly 6 times that of alfentanil or sufentanil. (Miller's Anesthesia 10e)
This means:
- Short cases: Fentanyl boluses are excellent - redistribution terminates effect rapidly, drug is predictable
- Long infusions (>2-3 hours): Fentanyl is a poor choice as a continuous infusion analgesic component - emergence will be delayed, respiratory depression unpredictable
- ICU sedation with fentanyl infusions causes significant drug accumulation; patients may remain obtunded for many hours after the infusion is stopped
7. CSHT DOES NOT EQUAL TIME TO RECOVERY
An important conceptual limitation: CSHT only describes a 50% fall in plasma concentration - it does not directly predict clinical recovery (awakening).
Recovery time also depends on:
a) The Margin Above the Awakening Threshold
- If a patient is maintained at a concentration far above the awakening concentration (e.g., for cardiac surgery), even a drug with a short CSHT will take longer to produce awakening
- If maintained just above the awakening threshold, the 50% CSHT will nearly equal the time to awakening (Fig 30-5, Barash)
b) Effect-Site Equilibration (t½ ke0)
- CSHT reflects plasma concentration, but awakening is determined by effect-site (brain) concentration
- There is always a time lag between plasma and brain concentration (the hysteresis loop)
- Fentanyl has a t½ ke0 of 6.4 minutes - slow plasma-to-brain equilibration
- Alfentanil has a t½ ke0 of 1.1 minutes - rapid equilibration
- Propofol has a short t½ ke0 - making it rapidly titratable
- For a brief single stimulus (e.g., intubation), alfentanil is logically superior to fentanyl as the EEG effect closely mirrors plasma levels
(Barash's Clinical Anesthesia 9e, p.2512)
c) Target for 50% vs. Target Needed for Recovery
- CSHT describes 50% reduction - but if 80% or 90% reduction is needed to cross the awakening threshold, CSHT underestimates recovery time
- "Context-sensitive time to recovery" is a related concept that maps the percentage decline needed for the specific clinical endpoint
8. REMIFENTANIL - THE CSHT IDEAL
Remifentanil deserves mention as the extreme opposite of fentanyl:
- Contains an ester linkage in its structure
- Metabolized by non-specific plasma and tissue esterases (not liver-dependent)
- CSHT is essentially constant and independent of infusion duration - approximately 3-4 minutes regardless of whether the infusion ran for 30 minutes or 10 hours
- This makes it ideal for prolonged surgery requiring rapid, predictable offset
- Major limitation: no residual analgesia after stopping infusion - multimodal analgesia must be planned preemptively
(Barash's Clinical Anesthesia 9e; Katzung 16e)
9. COMPARATIVE SUMMARY TABLE
| Property | Propofol | Fentanyl | Remifentanil |
|---|---|---|---|
| Mechanism of clearance | Hepatic (+ extrahepatic) glucuronidation | Hepatic CYP3A4 | Plasma esterases |
| Clearance rate | High (>1.5 L/min) | Moderate (~1 L/min) | Extremely high |
| Lipophilicity / Vdss | Moderate Vdss | Very large Vdss (~4-6 L/kg) | Small Vdss |
| CSHT after 1h infusion | ~10-15 min | ~50-60 min | ~3-4 min |
| CSHT after 8h infusion | ~30-40 min | ~280 min | ~3-4 min |
| CSHT duration-dependence | Minimal | Extreme | None |
| Effect-site t½ ke0 | Short (rapid onset/offset) | 6.4 min (slow) | Short |
| Clinical use for infusion | Excellent (TIVA/TCI) | Short boluses only | Ideal for infusion |
| Risk after prolonged infusion | Low | High (respiratory depression, delayed emergence) | None (no residual effect) |
10. CLINICAL PRACTICE IMPLICATIONS
For Propofol:
- TIVA: Safe for short and long cases; expected emergence is predictable regardless of case duration
- TCI (Target Controlled Infusion): Pharmacokinetic models (Marsh, Schnider) exploit the stable CSHT to allow effect-site targeting
- Day surgery / ambulatory anesthesia: Drug of choice due to rapid, predictable recovery
- Caution: Propofol has no analgesic properties; must always be combined with opioid or regional analgesia
For Fentanyl:
- Bolus dosing for short cases: Fentanyl remains an excellent opioid - a single 1-2 mcg/kg bolus gives 20-30 min analgesia via redistribution; duration predictable
- Avoid as continuous infusion for cases >1-2 hours: Use sufentanil, alfentanil, or preferably remifentanil
- ICU/long TIVA: Fentanyl infusions cause significant accumulation; plan for postoperative respiratory support
- Synergy with propofol: Fentanyl markedly reduces propofol CP50 (concentration needed to ablate response to skin incision and other stimuli), allowing lower propofol doses - this synergy is routinely exploited in TIVA
The "Balanced" Anesthesia Principle:
Combining propofol (favorable CSHT, hypnotic) with a short-acting opioid (favorable CSHT, analgesic) gives independent control of hypnosis and analgesia, with predictable recovery from both. The pharmacodynamic synergy between these two drugs is profound - reducing doses of each and minimizing side effects.
CITATIONS
- Barash's Clinical Anesthesia, 9th Edition - Context-Sensitive Half-Time; How Does CSHT Relate to Recovery?; Pharmacokinetics: General Principles for Intravenous Anesthetics
- Miller's Anesthesia, 10th Edition - Pharmacokinetics and Pharmacodynamics of Opioids; Total Intravenous Anesthesia with Propofol and Opioids
- Katzung's Basic and Clinical Pharmacology, 16th Edition - Pharmacokinetics (Anesthetics)
- Hughes MA, Glass PSA, Jacobs JR. Context-sensitive half-time in multicompartmental pharmacokinetic models for intravenous anesthetic drugs. Anesthesiology. 1992;76:334-341. (Original paper defining the concept)
Explain renal auto regulation and it’s relevance during preop fluid therapy 20 marks answer
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Loading Image
Reading File
Finding Sources
Reading File
Reading File
I now have comprehensive material from five authoritative textbooks and the key autoregulation diagram. I can now compose the complete 20-mark answer.
Renal Autoregulation and its Relevance During Perioperative Fluid Therapy
1. INTRODUCTION: WHY THE KIDNEY NEEDS AUTOREGULATION
The kidney filters approximately 180 L/day of plasma at a GFR of ~125 mL/min. Of this, 178.5 L is reabsorbed and only ~1.5 L excreted as urine. This means a small change in GFR produces a disproportionately large change in urine output. Without autoregulation, a 25% rise in blood pressure would increase GFR from 180 L/day to 225 L/day; if tubular reabsorption remained constant at 178.5 L/day, urine output would rise from 1.5 L/day to 46.5 L/day - a 30-fold increase that would rapidly deplete plasma volume. Autoregulation prevents this catastrophe.
The primary purpose of renal autoregulation is therefore not to maintain oxygen delivery (unlike in other organs) - it is to maintain a constant GFR to allow precise control of water and solute excretion. Normal resting renal blood flow (RBF) is approximately 1,200 mL/min (representing 20-25% of cardiac output), far exceeding the ~50-80 mL/min needed for metabolic needs alone.
(Guyton & Hall Medical Physiology; Brenner & Rector's The Kidney)
2. DEFINITION OF RENAL AUTOREGULATION
Renal autoregulation is the intrinsic ability of the kidney to maintain relatively constant renal blood flow (RBF) and glomerular filtration rate (GFR) despite changes in mean arterial pressure (MAP), independent of neural and hormonal influences.
Key features:
- Operates over a MAP range of approximately 70-180 mmHg (the autoregulatory plateau)
- Below MAP ~70 mmHg, autoregulation fails and RBF/GFR fall proportionally with pressure
- Above MAP ~180 mmHg, autoregulation is overwhelmed and flow rises
- A denervated (transplanted) kidney autoregulates as well as an intact kidney - confirming its intrinsic, non-neural nature
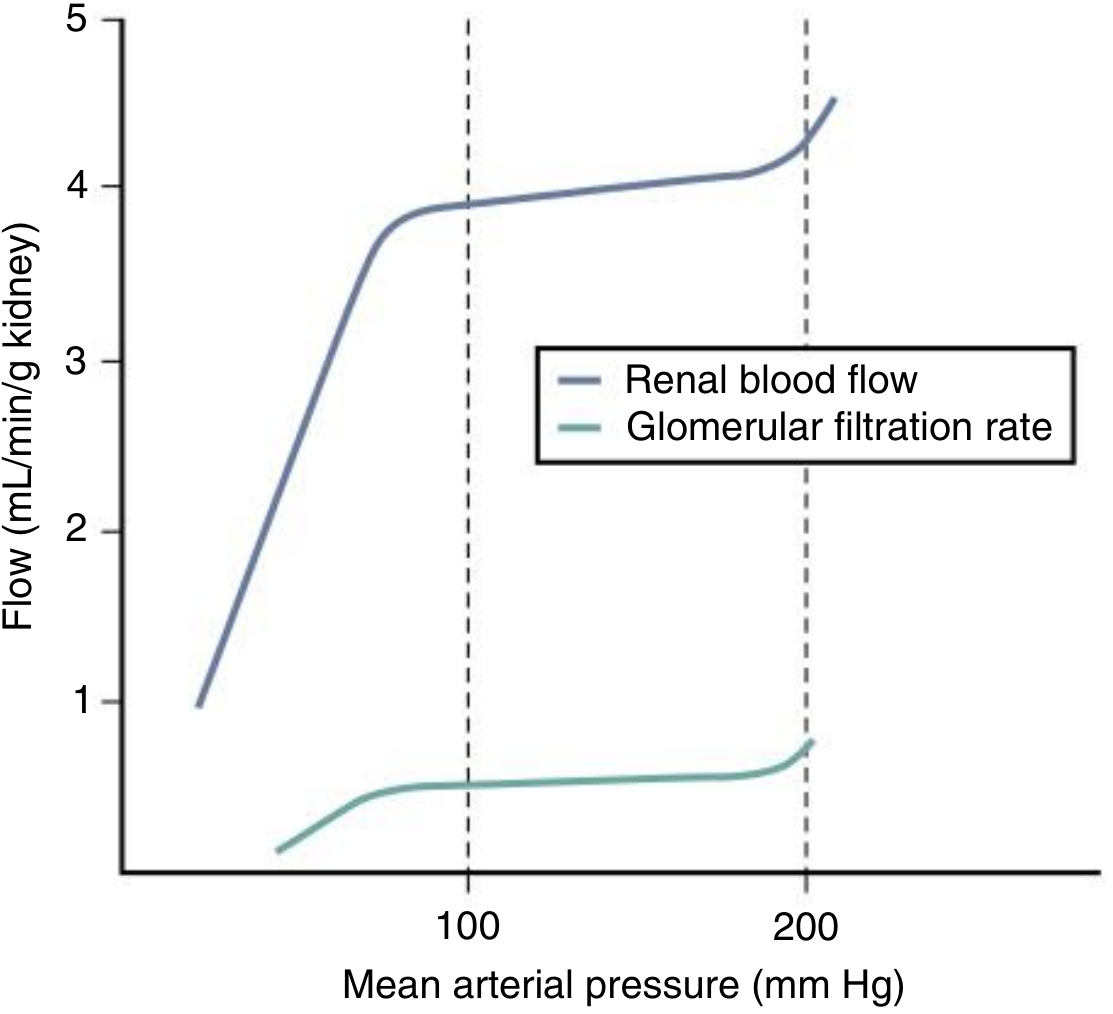
Fig: Renal Autoregulation of RBF and GFR. Between MAP 80-180 mmHg, both RBF and GFR remain relatively constant despite fluctuations in blood pressure. Below the lower limit, both fall precipitously. (Comprehensive Clinical Nephrology, 7e)
3. MECHANISMS OF RENAL AUTOREGULATION
Two principal mechanisms operate in parallel, acting mainly at the level of the afferent arteriole:
Mechanism 1: Myogenic Response (Fast - seconds)
Principle: Vascular smooth muscle responds directly to changes in wall tension (stretch).
Pathway:
- ↑ Renal perfusion pressure → stretches the wall of the afferent arteriole
- Mechanical stretch opens mechanosensitive (stretch-activated) cation channels → membrane depolarization
- Depolarization activates voltage-gated L-type calcium channels → Ca²⁺ influx into smooth muscle cells
- ↑ Intracellular Ca²⁺ → smooth muscle contraction → afferent arteriolar vasoconstriction
- ↑ Resistance offsets ↑ pressure → RBF remains constant
- Converse applies with decreased pressure: reduced stretch → smooth muscle relaxation → vasodilation → ↓ resistance
Speed: Responds within 3-10 seconds - suited for acute, transient changes in blood pressure (e.g., the pressor response to laryngoscopy)
Calcium channel blockade almost completely abolishes this mechanism - hence calcium channel blockers can impair renal autoregulation.
(Costanzo Physiology 7e; Brenner & Rector's The Kidney; Miller's Anesthesia 10e)
Mechanism 2: Tubuloglomerular Feedback (TGF) - Slower (20+ seconds)
Principle: The nephron monitors its own filtration rate and adjusts upstream blood flow via a feedback loop at the juxtaglomerular apparatus (JGA).
Anatomical substrate: The macula densa - a specialized group of cells in the thick ascending limb of the loop of Henle at the transition to the early distal tubule - lies in close anatomical proximity to its parent glomerulus and afferent/efferent arterioles. Together these form the juxtaglomerular apparatus.
Pathway when perfusion pressure rises:
- ↑ MAP → ↑ RBF → ↑ GFR → ↑ filtrate delivery to the thick ascending limb
- ↑ NaCl delivery to the macula densa
- Macula densa cells increase NaCl uptake via furosemide-sensitive Na⁺-K⁺-2Cl⁻ cotransporter (NKCC2)
- This triggers ATP release into the extracellular space
- ATP → metabolized to adenosine in the extracellular space → acts on adenosine A1 receptors on afferent arteriolar smooth muscle → vasoconstriction
- ↑ Afferent arteriolar resistance → ↓ glomerular hydrostatic pressure → ↓ GFR back to normal
Pathway when perfusion pressure falls:
- ↓ NaCl delivery → less macula densa activation → afferent arteriolar vasodilation + ↑ renin release → angiotensin II → efferent arteriolar constriction → maintains glomerular hydrostatic pressure → preserves GFR
TGF is modulated by:
- Angiotensin II: Sensitizes TGF (enhances its responsiveness)
- Nitric oxide (NO): Blunts TGF (vasodilatory counter-regulation); NO is not essential for TGF but modulates the plateau
- Prostaglandins: Vasodilatory prostaglandins (PGI2, PGE2) counteract excessive vasoconstriction - clinically relevant for NSAIDs
(Comprehensive Clinical Nephrology 7e; Costanzo Physiology 7e; Miller's Anesthesia 10e)
Mechanism 3: Glomerulotubular Balance (Supplementary)
Although not strictly autoregulation, glomerulotubular balance acts in concert with TGF:
- A constant fraction (~67%) of filtered water and solute is reabsorbed in the proximal tubule regardless of GFR
- This prevents the distal segments of the nephron from being overwhelmed during sudden GFR increases
- Protects distal tubular concentrating mechanisms even during rapid hemodynamic changes
(Miller's Anesthesia 10e)
4. LIMITS OF AUTOREGULATION AND WHEN IT FAILS
The autoregulatory plateau is not absolute. Several conditions shift the lower limit upward (meaning failure occurs at higher MAP than the usual ~70 mmHg):
| Condition | Effect on Autoregulation |
|---|---|
| Chronic hypertension | Lower limit shifts to higher MAP (~90-100 mmHg); kidney "adapted" to higher baseline pressure |
| Diabetic nephropathy | Impaired myogenic response; afferent arteriolar disease |
| CKD / renal artery stenosis | Narrow autoregulatory range; pressure-passive flow |
| Sepsis / SIRS | Endothelial dysfunction impairs NO-mediated and TGF mechanisms |
| NSAIDs / COX inhibitors | Block vasodilatory prostaglandins → afferent vasoconstriction → loss of lower-range buffering |
| ACE inhibitors / ARBs | Block angiotensin II → efferent arteriole dilates → cannot maintain glomerular pressure at low MAP |
| Severe volume depletion | RAAS activation initially compensatory; but severe Ang II drives afferent constriction and reduces GFR |
When MAP falls below the lower limit (~70 mmHg in normal individuals; higher in hypertensives), RBF and GFR fall in a pressure-passive manner, and ischemic acute tubular necrosis (ATN) may develop if hypoperfusion is severe or prolonged.
5. RENAL RESPONSE TO VOLUME DEPLETION
When hypovolemia reduces effective arterial blood volume, the kidney activates three major extrinsic responses that override intrinsic autoregulation in the direction of sodium and water conservation:
a) Sympathetic Nervous System Activation
- Renal sympathetic nerves are richly distributed to afferent and efferent arterioles and tubular cells
- Catecholamine release (especially norepinephrine) causes afferent arteriolar constriction - reducing RBF and GFR
- Also directly increases tubular sodium reabsorption via alpha-adrenergic receptors on tubular cells
- α2-adrenergic activity reduces the glomerular ultrafiltration coefficient
b) Renin-Angiotensin-Aldosterone System (RAAS)
- Hypovolemia → ↓ perfusion pressure at the JGA → ↑ renin release (from juxtaglomerular cells)
- ↓ NaCl delivery to macula densa → also triggers ↑ renin
- Renin → angiotensinogen → angiotensin I → ACE → angiotensin II
- Ang II preferentially constricts the efferent arteriole >> afferent arteriole at moderate volume depletion → maintains GFR (and glomerular hydrostatic pressure) despite reduced RBF (filtration fraction increases)
- In severe volume depletion, Ang II drives afferent constriction too → GFR falls
- Ang II also directly stimulates proximal tubular Na⁺-H⁺ exchangers → sodium reabsorption
- Ang II triggers aldosterone release → Na⁺ retention in collecting duct
c) Antidiuretic Hormone (AVP/ADH)
- Hypovolemia is a potent stimulus for AVP release (even overriding osmolarity signals)
- AVP causes: peripheral vasoconstriction + water reabsorption at collecting duct
- Urine becomes highly concentrated (osmolality up to 1200 mOsm/kg) with virtually no sodium (<10 mEq/L) - the hallmark of prerenal azotemia
Prerenal azotemia - rising BUN and creatinine with concentrated, sodium-poor urine - is the reversible early stage. If hypoperfusion continues, it progresses to ischemic ATN (acute tubular necrosis), which is slow to recover.
(Miller's Anesthesia 10e; Brenner & Rector's The Kidney)
6. PERIOPERATIVE FLUID THERAPY: THE CLINICAL RELEVANCE
Why the Perioperative Period is High Risk for Renal Autoregulatory Failure
Multiple factors in the surgical patient work together to challenge or overwhelm renal autoregulation:
| Factor | Mechanism of Renal Risk |
|---|---|
| Preoperative fasting (NPO) | Mild-moderate hypovolemia before incision |
| Bowel prep / diarrhea | Significant volume depletion; electrolyte loss |
| Anesthetic agents | Vasodilation (propofol, volatile agents) ↓ MAP; ↓ sympathetic tone |
| Neuraxial anesthesia | Sympathectomy → marked vasodilation → ↓ MAP |
| Surgical blood loss | ↓ effective circulating volume |
| IPPV / positive pressure ventilation | ↓ venous return → ↓ cardiac output → ↓ renal perfusion |
| Aortic cross-clamping | Direct ↓ renal perfusion pressure (especially infrarenal/suprarenal) |
| Intra-abdominal hypertension | ↑ renal vein pressure → ↓ net filtration pressure |
| NSAIDs (pre/postoperatively) | Block PGI2/PGE2 → loss of vasodilatory protection → afferent constriction |
| ACE inhibitors/ARBs (continued) | Cannot increase efferent resistance to maintain GFR when MAP drops |
| Sepsis/SIRS | Endothelial dysfunction, impaired TGF and NO signaling |
(Sabiston Textbook of Surgery; Miller's Anesthesia 10e; Brenner & Rector's The Kidney)
Principles of Perioperative Fluid Therapy Informed by Autoregulation
A. Maintain MAP Within the Autoregulatory Plateau
The single most important renal protection measure is maintaining MAP above the lower limit of autoregulation:
- In normal patients: keep MAP ≥65-70 mmHg
- In chronic hypertensives: their lower limit may be 90-100 mmHg; a MAP of 70 mmHg that seems "adequate" for normal patients may cause renal ischemia in them
- Clinical implication: Do not accept "permissive hypotension" in hypertensive patients or those with renal disease
B. Correct Preoperative Hypovolemia
Preoperative fasting (even modern 2-hour clear fluid fasting), bowel prep, fever, or medical illness can all reduce circulating volume before the first incision:
- Assess volume status before induction: history, urine output, skin turgor, HR, BP, dry mucous membranes
- Resuscitate with appropriate crystalloid or colloid before anesthesia where deficit is significant
- Correcting hypovolemia before vasodilation of anesthesia prevents the combined insult of low MAP + high RAAS/sympathetic tone on renal perfusion
C. Goal-Directed Fluid Therapy (GDFT)
Modern perioperative fluid management has moved away from both:
- Liberal fluid strategies (risk: interstitial edema, impaired bowel recovery, dilutional coagulopathy, pulmonary edema)
- Restrictive fluid strategies (risk: hypovolemia, renal hypoperfusion, AKI)
Toward goal-directed fluid therapy (GDFT), which titrates fluid to achieve specific hemodynamic endpoints:
- Stroke volume (SV) optimization via dynamic fluid responsiveness indices (pulse pressure variation, stroke volume variation, passive leg raise)
- Maintain cardiac output sufficient to meet end-organ demand
- Target urine output ≥0.5 mL/kg/hr as a crude index of renal perfusion (though intraoperative oliguria alone does not mandate aggressive fluid boluses - this remains debated)
D. The Importance of the "Two-Hit" Concept
Renal autoregulation can usually handle a single insult. Perioperative AKI usually requires two or more simultaneous insults:
- Hit 1: Hypovolemia + baseline CKD
- Hit 2: Intraoperative hypotension + nephrotoxic drug (aminoglycoside, contrast, NSAID)
- Hit 3: Sepsis + prolonged low MAP
This is why fluid therapy must be considered in the context of all concurrent renal stressors, not in isolation.
E. Specific Considerations for Medications
- NSAIDs: Block vasodilatory prostaglandins that normally counteract angiotensin II vasoconstriction at the afferent arteriole. In the volume-depleted patient with high RAAS activity, removing this prostaglandin protection unmasks unopposed vasoconstriction → precipitous ↓ GFR. Avoid or minimize perioperatively, especially in elderly, CKD, hypovolemic, or cardiac failure patients.
- ACE inhibitors/ARBs: In low-volume states, efferent arteriolar tone maintained by Ang II is the last defense of GFR. ACE-I/ARB blockade removes this protection. Consider holding on the day of major surgery (especially cardiac, aortic, or high-blood-loss procedures).
- Vasopressors: When hypotension is driven by vasodilation (neuraxial anesthesia, anesthetic agents), vasopressors (phenylephrine, norepinephrine) are preferable over excessive fluid to restore MAP and renal perfusion without the harms of fluid overload.
F. Markers of Adequacy of Renal Perfusion
| Marker | Interpretation |
|---|---|
| Urine output ≥0.5 mL/kg/hr | Suggests adequate perfusion (not perfect - oliguria can be appropriate hormonal response) |
| BUN:Creatinine ratio >20:1 | Suggests prerenal etiology (concentrated, sodium-poor urine) |
| Urine Na⁺ <20 mEq/L | Prerenal (RAAS-mediated sodium retention intact) |
| Urine osmolality >500 mOsm/kg | Concentrated urine = intact tubular function, prerenal pattern |
| Rising creatinine (>0.3 mg/dL within 48h) | AKI criterion (KDIGO) - requires urgent assessment and fluid optimization |
| Novel biomarkers: NGAL, KIM-1, IL-18 | Earlier markers of tubular injury, not yet routine |
7. HYPOVOLEMIA RESPONSE SUMMARY: SCHEMATIC
HYPOVOLEMIA / ↓ MAP
│
┌────┴────────────────────────────┐
│ │
Sympathetic activation RAAS activation
↓ RBF (afferent constriction) Ang II: efferent > afferent constriction
↑ Tubular Na reabsorption → GFR preserved initially
│ → Severe: GFR falls
└────────────────┬──────────────┘
│
AVP release
Water retention
Concentrated urine
│
[PRERENAL AZOTEMIA]
│
If not corrected:
[ISCHEMIC ATN - irreversible]
8. SPECIAL CLINICAL SCENARIOS
Chronic Hypertension
- Autoregulatory curve is right-shifted - lower limit now ~90-100 mmHg
- Seemingly "normal" MAP of 65-75 mmHg can cause renal ischemia
- Intraoperative MAP should ideally be maintained within 20% of the patient's baseline preoperative MAP
Diabetic Nephropathy
- Afferent arteriolar disease impairs myogenic response
- Glomerular hyperfiltration present at baseline; RAAS chronically activated
- These patients have very narrow autoregulatory range and are extremely sensitive to any perioperative hypotension
Cardiac Surgery (CPB)
- Non-pulsatile flow during cardiopulmonary bypass disrupts TGF signaling
- Hemodilution reduces viscosity (beneficial for flow) but also reduces oxygen carrying capacity
- Mean perfusion pressure on CPB should target ≥70-80 mmHg to maintain autoregulatory reserve
Septic AKI
- Endothelial NO dysregulation impairs autoregulation
- Microvascular injury causes heterogeneous cortical ischemia despite normal or elevated cardiac output
- Fluid therapy alone is insufficient; restoration of MAP with vasopressors is equally important
9. SUMMARY: KEY EXAMINATION POINTS
| Point | Detail |
|---|---|
| Autoregulatory range | MAP 70-180 mmHg (shifts rightward in hypertension) |
| Two mechanisms | Myogenic (fast, Ca²⁺ channels) + TGF (slow, adenosine/A1 receptor) |
| Primary purpose | Maintain constant GFR to regulate Na/H2O balance |
| Lower limit failure → | Prerenal → ATN continuum |
| Perioperative threats | Anesthetic vasodilation, hypovolemia, NSAID, RAAS blockade, sepsis |
| Fluid therapy goal | Restore MAP within autoregulatory plateau; target euvolemia not excess |
| GDFT | Dynamic indices (PPV, SVV) guide fluid vs vasopressor decision |
| Dangerous drugs | NSAIDs + ACEi/ARB in hypovolemic perioperative patient |
CITATIONS
- Miller's Anesthesia, 10th Edition - Renal Autoregulation; Renal Control of Intravascular Volume; Perioperative AKI
- Brenner and Rector's The Kidney - Intrinsic Mechanisms: Renal Autoregulation; Prerenal Acute Kidney Injury
- Comprehensive Clinical Nephrology, 7th Edition - Autoregulation of RBF and GFR; Renal Autoregulation
- Guyton & Hall Textbook of Medical Physiology - Autoregulation of GFR and Renal Blood Flow; Importance of GFR Autoregulation
- Costanzo Physiology, 7th Edition - Autoregulation of Renal Blood Flow
This is a shared conversation. Sign in to Orris to start your own chat.