klingetger syndrome in depth like as Paeditrician/ Neonatologist
Reading File
Finding Sources
Searching PubMed
Klinefelter syndrome 47XXY pediatric management
Reading File
Reading File
Finding Sources
Searching PubMed
Klinefelter syndrome 47XXY neonatal pediatric testosterone
Reading File
Reading File
Finding Sources
Searching PubMed
Klinefelter syndrome 47XXY diagnosis treatment review
Reading File
Finding Sources
Searching PubMed
"Klinefelter syndrome" testosterone replacement puberty
Loading Image
Loading Image
Reading File
Finding Sources
Reading File
Reading File
I now have comprehensive data from multiple authoritative textbooks. Let me compile the full in-depth response for a Paediatrician/Neonatologist.
Klinefelter Syndrome (47,XXY) - In-Depth for the Paediatrician/Neonatologist
1. Overview & Epidemiology
Klinefelter syndrome (KS) is the most common sex chromosome aneuploidy and the most common cause of primary hypogonadism in males.
| Parameter | Data |
|---|---|
| Incidence | ~1 in 600-660 live male births |
| Classic karyotype | 47,XXY (90% of cases) |
| Mosaic form | 46,XY/47,XXY (~15% of patients) |
| Diagnosis rate | Only ~25% diagnosed in lifetime (vast majority missed) |
By definition: any male with at least one Y chromosome and at least two X chromosomes has Klinefelter syndrome.
2. Genetics & Pathogenesis
Chromosomal origin
- Caused by non-disjunction of sex chromosomes during meiosis
- Maternal and paternal non-disjunction at meiosis I contribute roughly equally
- Advanced maternal age (>40 years) is a risk factor for maternal origin cases
- No phenotypic difference based on which parent provides the extra X
Why does an extra X cause hypogonadism? (Two mechanisms)
Mechanism 1 - Incomplete X-inactivation / gene dosage:
About 35% of X-linked genes escape Lyonization. These genes are therefore expressed at double dose in 47,XXY males compared to normal males - "overexpression" of one or more of these genes drives hypogonadism and somatic features.
Mechanism 2 - Androgen receptor CAG repeats:
The androgen receptor (AR) gene on the X chromosome contains polymorphic CAG trinucleotide repeats. Receptors with shorter CAG repeats are more sensitive to androgens. In XXY males, the X bearing the AR allele with the shortest repeat is preferentially inactivated - leaving the less sensitive long-repeat receptor dominant. This exacerbates hypogonadism and explains features like small penis size.
SHOX gene: Maps to the pseudoautosomal region of Xp, escapes X inactivation - the extra copy explains the tall stature and long legs characteristic of KS.
- Robbins & Kumar Pathologic Basis of Disease, p. 167
3. Clinical Presentation by Age
Prenatal / Prenatal Diagnosis
- The majority of KS cases diagnosed today come from prenatal chromosomal testing (amniocentesis for advanced maternal age, NIPT/cfDNA, or incidental finding on cytogenetics)
- No specific structural anomalies on fetal ultrasound reliably suggest KS
Neonatal Period (Birth to 1 month)
Key point for Neonatologists: KS is rarely clinically apparent at birth. The newborn examination is usually entirely normal.
Possible neonatal findings that may prompt suspicion:
- Micropenis - KS is among the causes of micropenis (deficient testosterone during fetal gonadal development)
- Small testes / undescended testes (cryptorchidism) - especially if bilateral
- Neonates with 47,XXY have a blunted minipuberty - the normal neonatal surge of LH, FSH, and testosterone (weeks 1-3 of life) is diminished. This window is a critical opportunity:
- If KS is already suspected (prenatal FISH/karyotype), check LH, FSH, testosterone, AMH, inhibin B at age 1-3 months during minipuberty
- Inhibin B and AMH are Sertoli cell markers - they are often already reduced in neonatal period, reflecting early germ cell loss
- Chromosome analysis (karyotype or chromosomal microarray) confirms diagnosis
Infancy & Early Childhood (1 month - 5 years)
- Mostly asymptomatic
- Developmental milestones may be subtly delayed - especially speech and language (this is often the first clinical clue in a child with prenatal diagnosis)
- Motor development can be slightly delayed
- Hypotonia may be mild
- No distinctive dysmorphic features in most children; prepubertal boys appear physically normal with the exception of disproportionately long legs
School Age (5-12 years)
- Tall stature with long legs (due to extra SHOX copy) - pediatrician may note height above expected family percentile
- Learning difficulties - predominantly in:
- Reading and language comprehension (verbal IQ typically lower than performance IQ)
- Executive function
- Language difficulties lead to shyness, unassertiveness, social withdrawal, increased risk of depression
- Educational support and speech therapy may be needed
- Attention deficit and behavioral difficulties are more common than in the general population
- Testes remain prepubertal in size but are otherwise not grossly abnormal
Puberty (12-18 years) - When Classic Features Emerge
This is the age when KS is most commonly first diagnosed clinically.
| Feature | Notes |
|---|---|
| Puberty onset | Normal timing - puberty begins on schedule |
| Testes | Remain small and firm - do not grow to adult size (typically <3.5 cm in length / <6 mL volume by orchidometer); hallmark finding |
| Gynecomastia | Present in ~40-50%; due to elevated estradiol:testosterone ratio |
| Body habitus | Eunuchoid - tall with long legs, reduced muscle mass, female-type fat distribution |
| Penis | May be small; inadequate virilization |
| Secondary sexual characteristics | Reduced facial hair, sparse axillary hair; pubic hair usually present |
| Libido/erectile function | Often impaired in adolescence/adulthood |
| Cognitive/behavioral | Verbal learning difficulties, executive function deficits, depression risk |
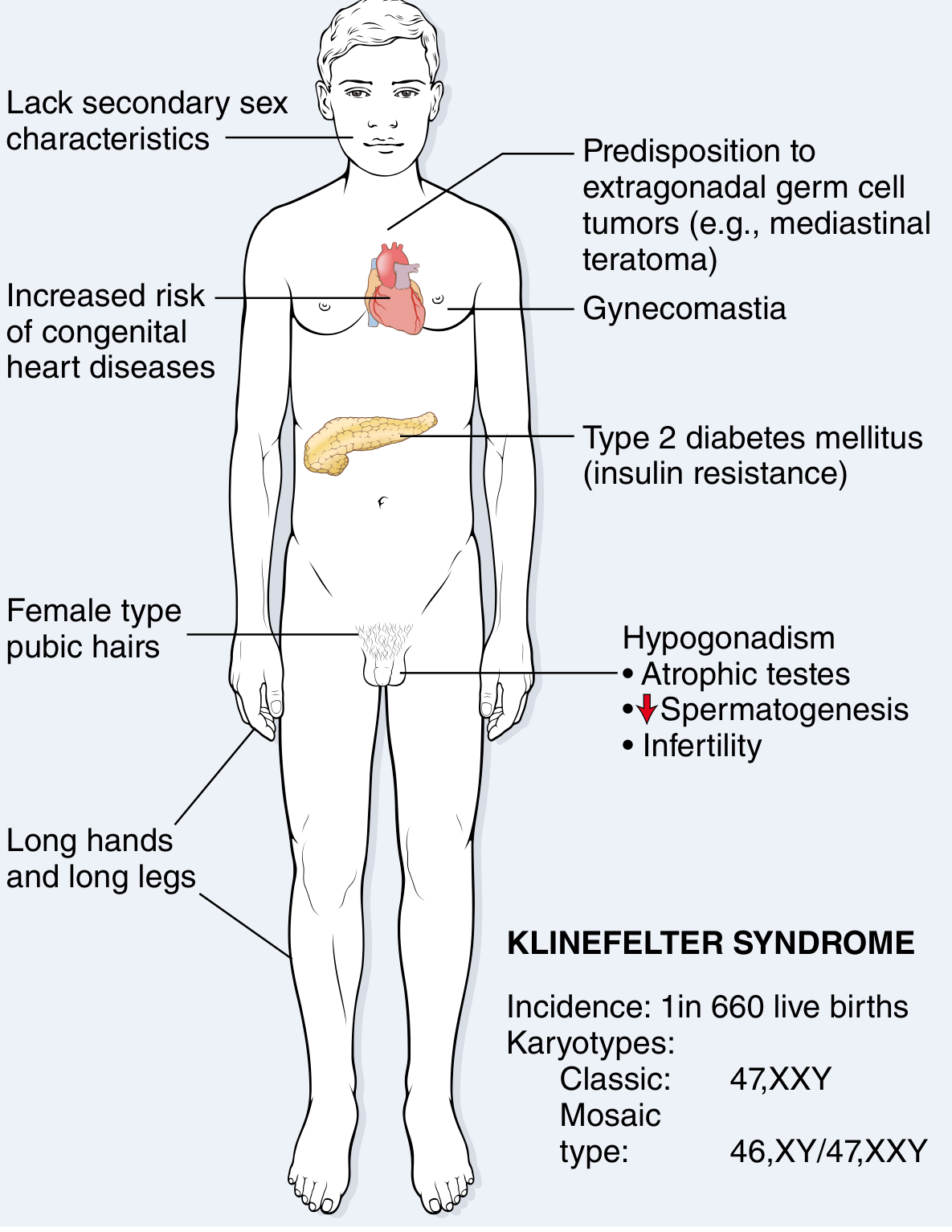
Clinical features of Klinefelter syndrome (47,XXY). Incidence: 1 in 660 live births - Robbins & Kumar Pathologic Basis of Disease
4. Phenotype of Classic 47,XXY vs Mosaic vs Variants
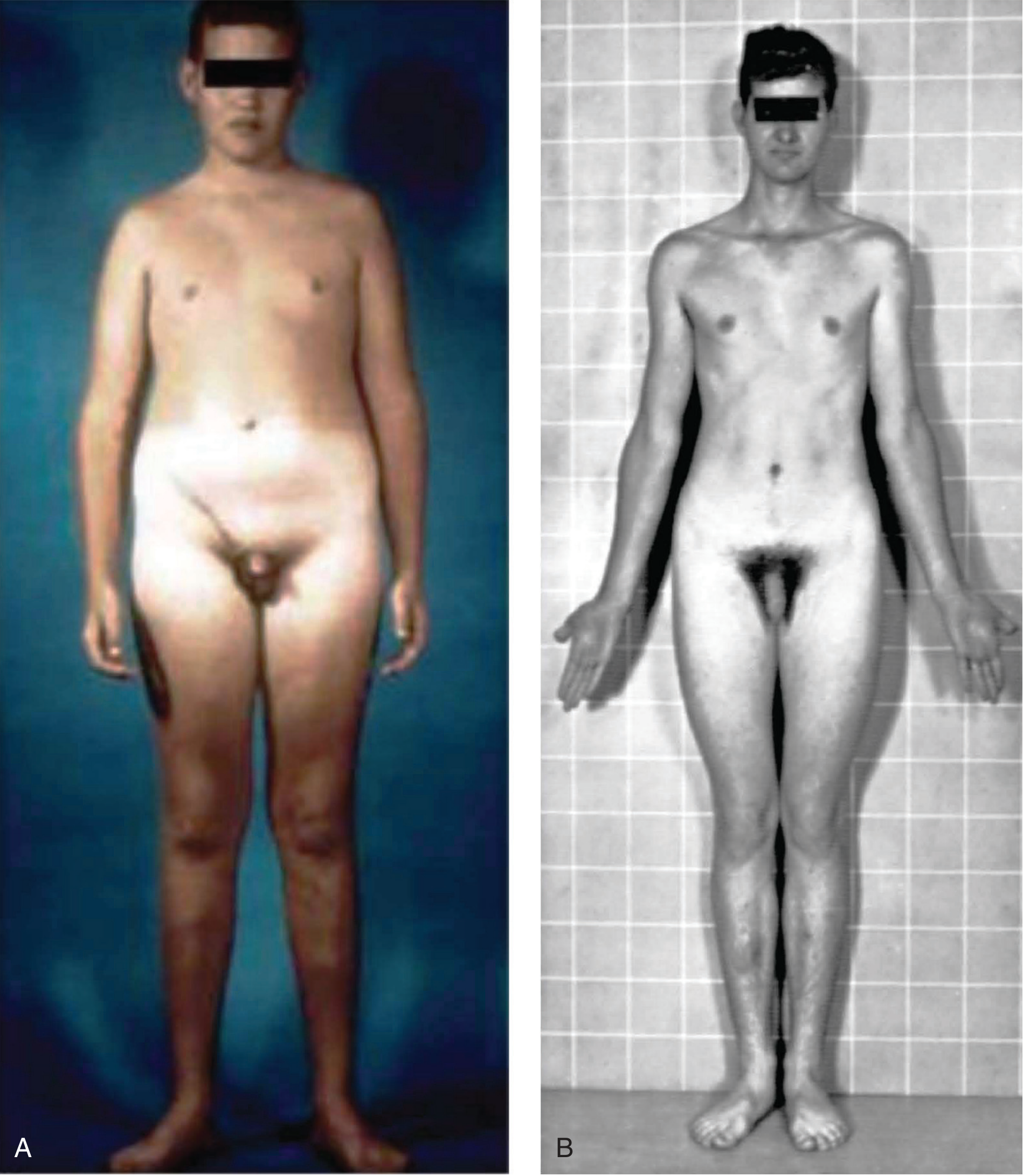
Phenotype of 47,XXY - patients appear physically normal until puberty when signs of hypogonadism become obvious - Thompson & Thompson Genetics, p.122
Karyotype Variants
| Karyotype | Incidence | Features |
|---|---|---|
| 47,XXY | 1/650 males | Classic KS - as described above |
| 46,XY/47,XXY (mosaic) | ~15% of KS | Milder features; some may have near-normal testicular function; occasional spermatogenesis; fertility possible without ART |
| 48,XXXY | 1/25,000 males | More severe phenotype; intellectual disability more likely; additional dysmorphic features |
| 48,XXYY | 1/10,000 males | Tall stature; vascular anomalies (angiomas, acrocyanosis, peripheral vascular disease), hypertelorism |
| 49,XXXXY | ~1/85,000 males | Severe intellectual disability; radial-ulnar synostosis; congenital heart disease; marked facial dysmorphism; genital abnormalities |
General rule: The more X chromosomes, the more severe the phenotype, and the greater the likelihood of intellectual disability.
5. Testicular Pathology
The progressive loss of germ cells is the central pathological process:
- Seminiferous tubules undergo progressive hyalinization and fibrosis - replaced by pink collagenous "ghosts"
- Some tubules remain embryonic (cords without lumens, never progressing to mature spermatogenesis)
- Leydig cells appear prominent (due to crowding from tubule atrophy and elevated gonadotropins) - but their absolute volume is actually normal or reduced
- Germ cell depletion begins at puberty - spermatogonial stem cells with 47,XXY are progressively lost
- Some patients still have focal spermatogenesis - basis for micro-TESE fertility
Hormonal Pattern
| Hormone | Level |
|---|---|
| FSH | Consistently elevated (most reliable marker) |
| LH | Elevated |
| Testosterone | Variably decreased (often low-normal in adolescence, falling further with age) |
| Estradiol | Elevated (mechanism unclear) |
| Inhibin B / AMH | Low (Sertoli cell dysfunction) |
Elevated FSH + elevated LH + low testosterone = hypergonadotropic hypogonadism
The elevated estradiol:testosterone ratio drives gynecomastia and feminization.
- Robbins & Kumar Pathologic Basis of Disease, p. 167; Campbell Walsh Wein Urology
6. Comorbidities - What Every Paediatrician Must Monitor
Metabolic & Cardiovascular
- Type 2 diabetes / metabolic syndrome / insulin resistance - significantly higher risk
- Obesity - central adiposity, higher BMI
- Congenital heart disease - mitral valve prolapse (~50% of adults), ASD, VSD
- Osteoporosis / fractures - from testosterone deficiency → reduced bone mineral density; monitor from adolescence
- Thromboembolic disease - venous thrombosis risk increased
Oncologic
- Extragonadal germ cell tumors - 20-30-fold higher risk; especially mediastinal teratoma/NSGCT - check mediastinum in any young male with 47,XXY and chest symptoms
- Breast cancer - 8x higher risk than normal males; periodic mammography recommended
- Non-Hodgkin's lymphoma - increased risk
- Paradoxically, reduced risk of prostate cancer
Autoimmune
- Systemic lupus erythematosus (SLE) - significantly higher prevalence
- Sjögren's syndrome
- Other autoimmune diseases
Neuropsychiatric
- Learning disabilities (verbal IQ < performance IQ)
- Speech and language delay
- Executive dysfunction
- Depression and anxiety
- Autism spectrum features (more frequent in higher-order aneuploidies)
7. Diagnosis
When to Suspect and Test
Neonatal clues:
- Prenatal diagnosis on amniocentesis or NIPT
- Micropenis or bilateral cryptorchidism (especially if associated)
- Small/undescended testes
Childhood clues:
- Tall stature (above family expected)
- Speech and language delay
- Learning difficulties (especially reading)
- Behavioral difficulties / ADHD features
Pubertal clues (most common time of diagnosis):
- Small, firm, non-enlarging testes after onset of puberty
- Gynecomastia
- Eunuchoid habitus
- Delayed/incomplete virilization
- Elevated FSH
Adult clues:
- Infertility / azoospermia
- Hypogonadism symptoms
Investigations
| Investigation | Finding |
|---|---|
| Karyotype (gold standard) | 47,XXY or mosaic pattern |
| Chromosomal Microarray | Can detect mosaicism better than standard karyotype |
| FISH | Rapid confirmation, especially prenatal |
| FSH, LH | Elevated (even prepubertally in confirmed cases) |
| Testosterone | Low-normal → low |
| Estradiol | Elevated |
| Inhibin B / AMH | Low (Sertoli cell markers - most informative in neonatal/prepubertal period) |
| Semen analysis | Azoospermia (rule in adulthood; oligospermia suggests mosaicism) |
| Bone density (DEXA) | Reduced BMD - monitor from adolescence |
8. Management - Pediatric & Adolescent Focus
Neonatal Period
If prenatally diagnosed:
- Refer to pediatric endocrinology early
- Check minipuberty hormones at 1-3 months (LH, FSH, testosterone, inhibin B, AMH)
- Assess for micropenis/cryptorchidism - refer to pediatric urology if present
- Counselling - parents need to understand the wide phenotypic spectrum and that many boys lead essentially normal lives
Early Childhood (0-5 years)
- Speech and language therapy - should be initiated early, even before formal delay is documented, in all diagnosed boys
- Early intervention programs for developmental support
- Monitor growth - measure height, weight regularly
- Audiology assessment if indicated
School Age (5-12 years)
- Formal neuropsychological testing - assess verbal IQ, executive function, reading
- Educational support - individualized educational plans where needed
- Monitor for behavioral difficulties, ADHD, social difficulties
- Psychological support if needed
Puberty (12-16+ years)
Testosterone Replacement Therapy (TRT) - the cornerstone of pubertal management
- When to start: When signs of pubertal delay/arrest or hypogonadism appear - typically at age 11-14 years, guided by pubertal staging, bone age, and hormone levels
- Goal: Achieve normal virilization - voice deepening, penile growth, muscle development, axillary/facial hair, normal libido
- Important note before starting TRT: Offer fertility counselling. Once exogenous testosterone is started, suppression of residual spermatogenesis may occur. Sperm retrieval (micro-TESE) should be discussed - though evidence suggests retrieval rates in adolescence are similar to adulthood
TRT formulations in adolescents:
- IM testosterone enanthate/cypionate - low dose initially, escalating over 2-3 years to adult doses
- Transdermal gels increasingly preferred (more physiological delivery)
- Avoid supraphysiological levels
Gynecomastia:
- If significant, does not usually regress with TRT alone
- Reduction mammoplasty may be needed for severe cosmetic/psychological impact
Bone health:
- Monitor bone mineral density (DEXA scan) from mid-adolescence
- Ensure adequate calcium and vitamin D intake
- TRT reduces fracture risk by improving BMD
Fertility
- The majority of 47,XXY males are azoospermic - azoospermia is the rule
- Micro-TESE (microdissection testicular sperm extraction) - sperm retrieval rates 40-50%; higher in younger men
- Retrieved sperm used with ICSI - meta-analysis shows 43% live birth rate per ICSI cycle
- Mosaic 46,XY/47,XXY men may have sperm in ejaculate
- Preimplantation genetic testing (PGT) is sometimes used but not mandatory - >200 normal live births reported with ICSI alone
Surveillance (Adolescent and Transition to Adult Care)
| Organ System | Monitoring |
|---|---|
| Breast | Periodic breast self-examination; mammography in adulthood |
| Testes | Regular testicular examination for Leydig/Sertoli cell tumors and extragonadal GCT |
| Mediastinum | Awareness of extragonadal GCT risk - chest imaging if symptoms |
| Metabolic | Fasting glucose, lipids, BMI annually from adolescence |
| Bone | DEXA scan at initiation of TRT and periodically thereafter |
| Cardiac | Echocardiography at diagnosis (MVP, ASD, VSD) |
| Thyroid | Autoimmune thyroid disease surveillance |
| Mental health | Regular screening for depression, anxiety |
9. Neurodevelopment - Special Emphasis for the Paediatrician
KS has a significant neurodevelopmental profile that varies among individuals:
- Language delay is the most consistent finding - often first sign in a prenatally diagnosed child
- Verbal IQ is typically 10-15 points below performance IQ; full-scale IQ is usually within the normal range (not intellectually disabled in classic 47,XXY)
- Deficits in reading, language comprehension, verbal memory
- Executive dysfunction - planning, organization, cognitive flexibility
- Neuroimaging: Selective volume reductions in frontal and temporal regions (corresponding to language/executive areas)
- Psychosocial: Shyness, unassertiveness, apparent immaturity, higher rates of depression
- Higher X polysomies (48,XXXY; 49,XXXXY) - intellectual disability becomes more likely and more severe with each additional X
Early speech therapy and educational intervention are the single most important modifiable interventions in childhood.
10. Key Points Summary for Clinical Practice
| Age | Action |
|---|---|
| Prenatal | If NIPT/amnio shows 47,XXY - detailed counselling, plan neonatal evaluation |
| Newborn | Examine genitalia; if micropenis/cryptorchidism - check karyotype; plan minipuberty hormone evaluation at 1-3 months |
| 0-5 years | Speech therapy, developmental monitoring, parent education |
| 5-12 years | Neuropsychological testing, school support, monitor growth |
| Puberty | Testosterone therapy - start at evidence of hypogonadism; fertility counselling first |
| All ages | Monitor for comorbidities: bone density, cardiac, metabolic, oncologic, mental health |
Recent Evidence (PubMed, 2022-2025)
- The TIPY Study (PMID: 40090688) is an ongoing RCT examining the effect of testosterone treatment during puberty in boys with KS - results are awaited and may define optimal timing and dosing
- Pozza et al. 2023 (PMID: 37043499) documented testicular dysfunction beginning as early as infancy in 47,XXY boys, reinforcing the importance of neonatal evaluation during minipuberty
- López Krabbe et al. 2023 (PMID: 37010084) examined bone mineral content and body composition in boys and adolescents with KS on testosterone therapy
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 167
- Harrison's Principles of Internal Medicine 22E (2025), Chap. 402-403
- Campbell Walsh Wein Urology, 3-Volume Set, p. 1283-1284
- Thompson & Thompson Genetics and Genomics in Medicine, 9th ed., p. 122
- Langman's Medical Embryology
- Guyton and Hall Medical Physiology
This is a shared conversation. Sign in to Orris to start your own chat.