Interstitial lung disease..honey comb lung
Reading File
Finding Sources
Finding Sources
Searching PubMed
"interstitial lung disease" AND "honeycombing"
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
honeycomb lung HRCT interstitial pulmonary fibrosis
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Here is a comprehensive overview of Interstitial Lung Disease (ILD) and Honeycomb Lung, drawn from authoritative textbooks.
Interstitial Lung Disease (ILD) & Honeycomb Lung
What is ILD?
Interstitial lung disease (ILD) — also called diffuse parenchymal lung disease (DPLD) — is a heterogeneous group of disorders characterized predominantly by inflammation and fibrosis of the lung interstitium, producing a restrictive pattern on pulmonary function tests.
Major Categories (Robbins Pathologic Basis of Disease)
| Category | Examples |
|---|---|
| Fibrosing | IPF (UIP), NSIP, COP, connective tissue disease–associated, pneumoconiosis, drug reactions, radiation |
| Granulomatous | Sarcoidosis, hypersensitivity pneumonitis |
| Eosinophilic | Eosinophilic pneumonia |
| Smoking-related | DIP, RB-ILD |
| Other | Langerhans cell histiocytosis, PAP, LIP |
Common Clinical Features
- Progressive dyspnea and dry cough
- End-inspiratory crackles (Velcro crackles)
- Tachypnea, eventual cyanosis and clubbing
- Restrictive defect on PFTs: ↓ FVC, ↓ TLC, ↓ DLCO
- Chest imaging: bilateral nodules, reticular lines, ground-glass opacities
Honeycomb Lung — Definition
The Fleischner Society defines honeycombing as:
"The appearance of clustered cystic air spaces, typically of comparable diameters on the order of 3–10 mm (occasionally up to 2.5 cm), usually subpleural, with well-defined walls."
It represents established, irreversible pulmonary fibrosis — the final common pathway of many ILDs, referred to as end-stage lung or honeycomb lung.
The Prototypical Cause: Idiopathic Pulmonary Fibrosis (IPF)
IPF is the most important cause of honeycomb lung. Its histologic pattern is Usual Interstitial Pneumonia (UIP).
Pathogenesis
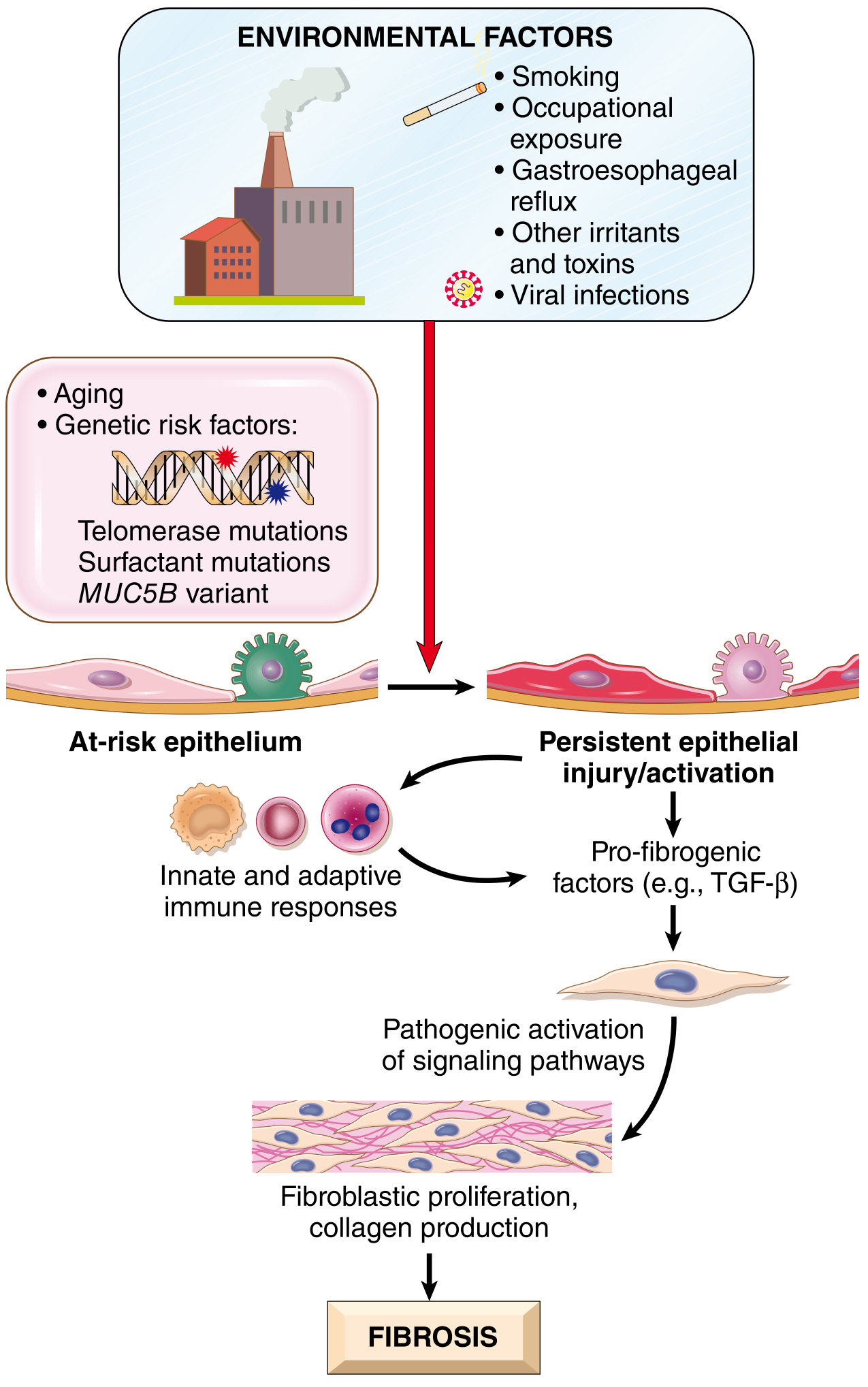
Proposed pathogenesis of IPF: recurrent alveolar epithelial injury in genetically susceptible individuals triggers TGF-β–driven fibroblast activation and collagen deposition.
Key factors:
- Environmental: Cigarette smoking (OR 1.6–9.4), metal/wood dust, microaspiration, air pollution, farming
- Genetic: Mutations in TERT, TERC, PARN, RTEL1 (telomere maintenance); MUC5B promoter polymorphism (↑risk); surfactant gene mutations in familial forms
- Age: Rarely before age 50; peak at 55–75 years
Histopathology
Gross
- Pleural surfaces are cobblestoned due to interlobular septal scarring
- Cut surface: firm, rubbery white fibrosis — lower lobe, subpleural, and periseptal distribution
Microscopic (UIP Pattern)
Hallmarks:
- Spatial heterogeneity — normal lung alternating with architecturally effaced lung at low power
- Temporal heterogeneity — early fibroblastic foci (loose myxoid stroma) alongside dense mature fibrosis
- Honeycomb fibrosis — destruction of alveolar architecture forming cystic spaces lined by metaplastic/hyperplastic type II pneumocytes or bronchiolar epithelium
- Mild lymphocytic inflammation; smooth muscle hyperplasia; pulmonary arterial hypertensive changes
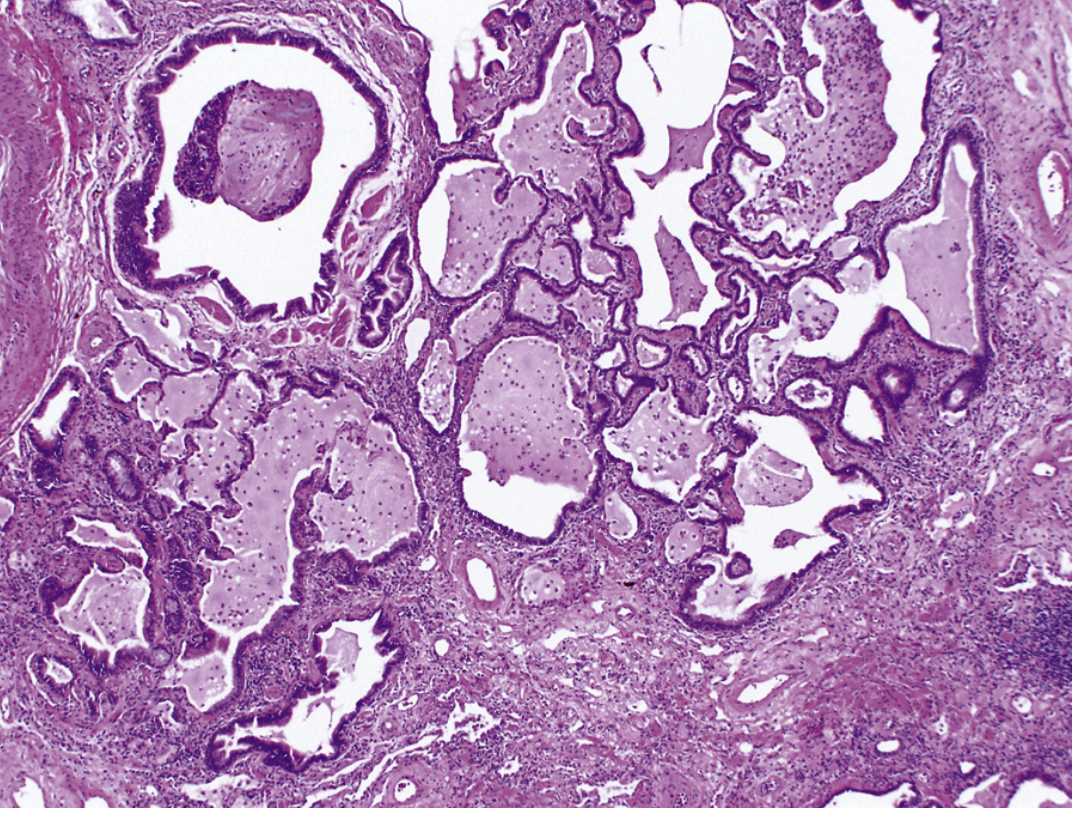
Fig. 89.5 — Honeycomb lung in IPF. Dense fibrous bands destroy alveolar architecture, forming cystic spaces lined by metaplastic epithelium. (×10, Murray & Nadel's Textbook of Respiratory Medicine)
Radiology — HRCT Features
HRCT is the cornerstone of diagnosis and has largely replaced open lung biopsy in typical cases.
HRCT findings in UIP/IPF
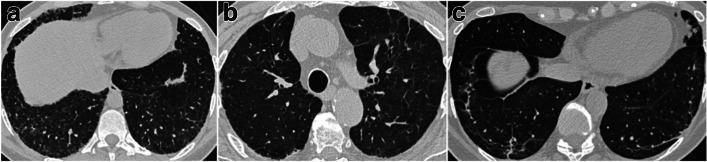
HRCT patterns in ILD: (a) honeycombing — subpleural clustered cysts with thick shared walls; (b) traction bronchiectasis — dilated, distorted bronchi within fibrotic lung; (c) architectural distortion.
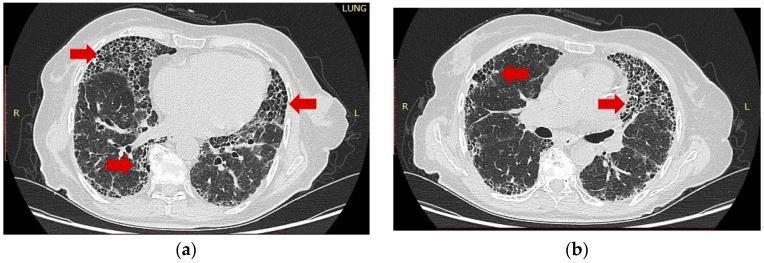
Bilateral, peripheral, basal-predominant honeycombing with traction bronchiectasis — hallmark UIP pattern on HRCT.
| Feature | Honeycombing | Mimics to Exclude |
|---|---|---|
| Location | Subpleural, basal-predominant | — |
| Cyst size | 3–10 mm (up to 2.5 cm) | — |
| Walls | Thick, well-defined, shared (stacked layers) | Paraseptal emphysema (thinner walls, no sharing) |
| Associated findings | Traction bronchiectasis, reticulation, architectural distortion | Cystic lung disease (LAM, LCH — no fibrosis context) |
Gross lung sections showing honeycomb pattern:
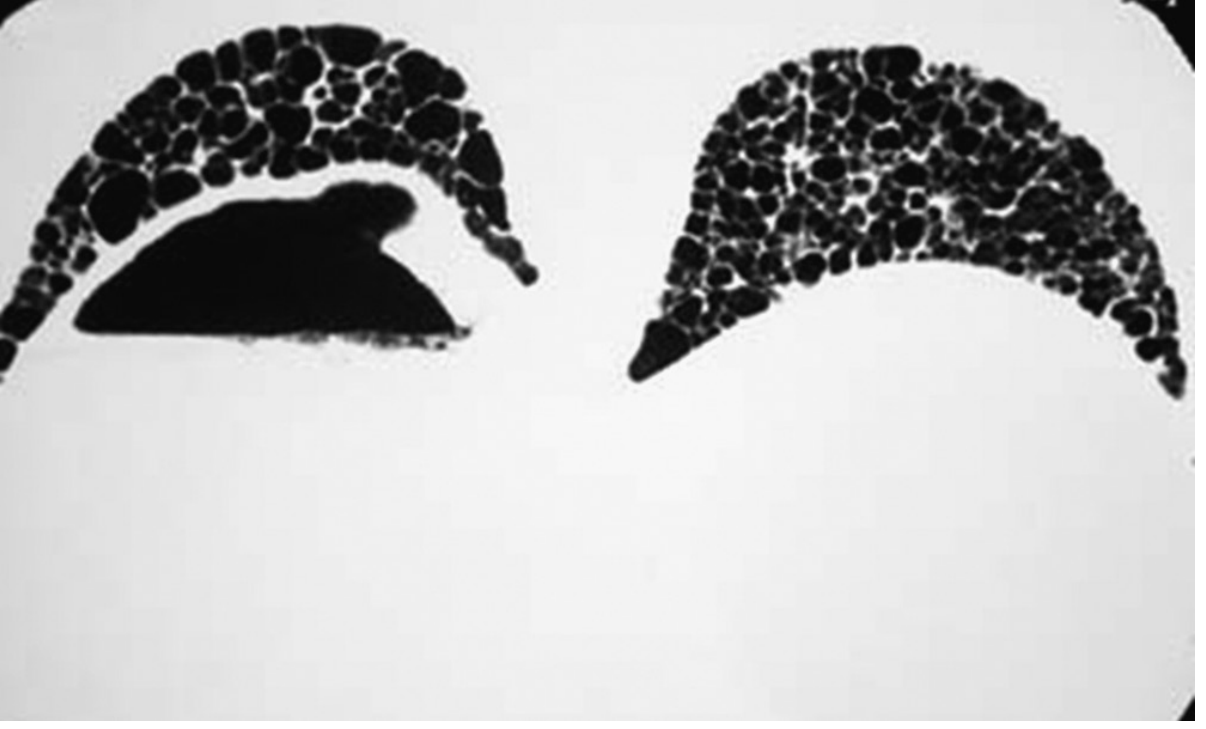
Cross-sections of the lung bases showing bilateral honeycombing in UIP/IPF — note clustered, thick-walled cystic spaces forming the "honeycomb" appearance.
Key distinction: In NSIP (Non-Specific Interstitial Pneumonia), ground-glass opacities do not progress to honeycombing (ground-glass → fibrosis stays as fine reticulation). Progression of ground-glass to honeycombing points to UIP and indicates irreversible fibrosis.
Clinical Course & Prognosis
- Median survival: ~3.8 years after IPF diagnosis
- Course is unpredictable — usually slow progressive respiratory failure; some patients have acute exacerbations (DAD superimposed on chronic UIP) with rapid deterioration
- Late complications: Pulmonary hypertension and cor pulmonale (right heart failure)
- Clubbing and cyanosis occur late
Treatment
| Intervention | Notes |
|---|---|
| Nintedanib (tyrosine kinase inhibitor) | Slows disease progression in IPF |
| Pirfenidone (TGF-β antagonist) | Slows disease progression in IPF |
| Lung transplantation | Only definitive therapy |
| Treat underlying cause | For secondary ILDs (stop smoking, treat CTD, remove antigen in HP) |
| Immunosuppressants | Effective in NSIP; not in IPF |
Prognosis is markedly better if patients completely cease smoking (e.g., in smoking-related ILD); end-stage honeycomb lung rarely develops after complete smoking cessation.
Key Summary
| Aspect | Key Point |
|---|---|
| Honeycomb lung | End-stage, irreversible fibrosis — final common pathway of many ILDs |
| Prototype | IPF (UIP pattern) |
| Histology | Cystic spaces lined by metaplastic epithelium + spatial/temporal heterogeneity |
| HRCT | Subpleural, basal-predominant clustered cysts 3–10 mm, thick shared walls |
| Pathogenesis | Aberrant epithelial repair → TGF-β → fibroblast activation → collagen deposition |
| Treatment | Nintedanib, pirfenidone (slow progression); transplant (definitive) |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Murray & Nadel's Textbook of Respiratory Medicine; Goldman-Cecil Medicine; Fishman's Pulmonary Diseases and Disorders
Can u explain in understandBLE TERMS
Interstitial Lung Disease & Honeycomb Lung — In Simple Terms
🫁 First, How Normal Lungs Work
Think of your lungs like a sponge — millions of tiny air sacs (alveoli) surrounded by thin walls. When you breathe in, these sacs fill with air, oxygen passes through the thin walls into your blood, and CO₂ comes back out.
The "interstitium" is just the name for those thin walls and the tissue between the air sacs.
What Goes Wrong in ILD?
In Interstitial Lung Disease, those thin walls get inflamed and then scarred (fibrosed).
Scar tissue is stiff. So instead of a soft, stretchy sponge, the lungs become like a stiff, rigid sponge that can't expand properly.
This is why patients feel:
- 😮💨 Breathlessness — the lungs can't stretch to take in a full breath
- 🤧 Dry cough — irritation from the scarring
- 😴 Fatigue — body isn't getting enough oxygen
What is "Honeycomb Lung"?
When the scarring gets very advanced, the normal air sac architecture is completely destroyed. What's left is a bunch of small, air-filled cysts (bubble-like holes) separated by thick fibrous walls.
On a CT scan or when you look at the actual lung, it looks exactly like a honeycomb — rows of small round holes with thick walls between them.
🍯 Honeycomb lung = end-stage scarring. It means the damage is permanent and cannot be reversed.
The Most Common Cause — IPF
IPF (Idiopathic Pulmonary Fibrosis) is the #1 cause of honeycomb lung.
- Idiopathic = we don't know the exact cause
- Pulmonary fibrosis = scarring of the lungs
Why does it happen?
Think of it like a wound that never heals properly:
- Something injures the lung lining (smoking, dust, pollution, aging)
- The body tries to repair it — sends in "repair cells" (fibroblasts)
- In IPF, those repair cells go into overdrive and lay down too much scar tissue
- This keeps happening again and again → progressive scarring → honeycomb lung
Who gets it?
- Usually people aged 55–75 years
- More common in smokers (risk increased up to 9x)
- Workers exposed to metal dust, wood dust, farming
What Does It Look Like Under the Microscope?
| What you see | What it means |
|---|---|
| Patches of normal lung next to destroyed lung | Damage is uneven — some areas worse than others |
| Young scar tissue alongside old scar tissue | Injury keeps happening repeatedly over time |
| Cystic holes lined by the wrong type of cells | The original air sac cells are replaced by scar and abnormal cells |
How is it Diagnosed?
HRCT Scan (High-Resolution CT)
This is the main tool. Doctors look for:
| Finding | What it looks like | What it means |
|---|---|---|
| Honeycombing | Clustered small holes (3–10 mm) at the edges of the lungs | Advanced irreversible fibrosis |
| Traction bronchiectasis | Airways stretched and distorted | Surrounding scar is pulling them apart |
| Reticulation | Net-like pattern | Thickened scarred walls between air sacs |
| Ground-glass opacity | Hazy, misty areas | Early inflammation (can be reversible) |
📍 In IPF, these changes are worst at the lower lobes and the outer edges (subpleural) of the lung — this distribution is a key clue.
How Does It Progress?
Normal Lung
↓ (repeated injury)
Inflammation
↓ (failed repair)
Scarring (fibrosis)
↓ (progressive)
Honeycomb Lung (end-stage)
↓
Respiratory failure + Pulmonary hypertension
- Most patients slowly get worse over years
- Some suddenly deteriorate (acute exacerbation)
- Average survival after IPF diagnosis: ~3.8 years
- Late features: blue lips/fingers (cyanosis), finger clubbing, right heart failure
Treatment — Can it Be Fixed?
Unfortunately, honeycomb lung cannot be reversed. But:
| Treatment | What it does |
|---|---|
| Nintedanib (drug) | Blocks signals that tell fibroblasts to keep scarring → slows progression |
| Pirfenidone (drug) | Blocks TGF-β, a key "scar more" signal → slows progression |
| Stop smoking | The single most important thing — dramatically slows progression |
| Lung transplant | The only cure, but not available to everyone |
These drugs slow the disease down — they don't cure it or reverse the damage already done.
Quick Summary
| Question | Simple Answer |
|---|---|
| What is ILD? | Scarring of the walls between lung air sacs |
| What is honeycomb lung? | End-stage scarring — looks like a honeycomb on CT |
| What causes it? | Most often IPF — repeated injury + failed repair |
| Who is at risk? | Smokers, older adults, dust-exposed workers |
| Is it reversible? | No — once honeycombed, that damage is permanent |
| What can we do? | Slow it down with drugs; transplant is the only cure |
Pneumoconiosis lung picture
Reading File
Finding Sources
Searching Images
pneumoconiosis silicosis coal worker lung pathology histology
Reading File
Loading Image
Loading Image
Finding Sources
Loading Image
Loading Image
Reading File
Searching Images
silicosis coal workers pneumoconiosis gross pathology black lung nodules histology
Loading Image
Loading Image
Loading Image
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.