M band other differential except multiple myeloma
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Here is the complete, textbook-sourced answer:
M Band (Monoclonal Protein) - Differential Diagnosis Other Than Multiple Myeloma
The M protein (paraprotein) on serum protein electrophoresis is NOT synonymous with multiple myeloma. It is found across a wide spectrum of plasma cell and lymphoproliferative disorders. The chart below from the Mayo Clinic series (Goldman-Cecil Medicine) shows the distribution when an M band is discovered:
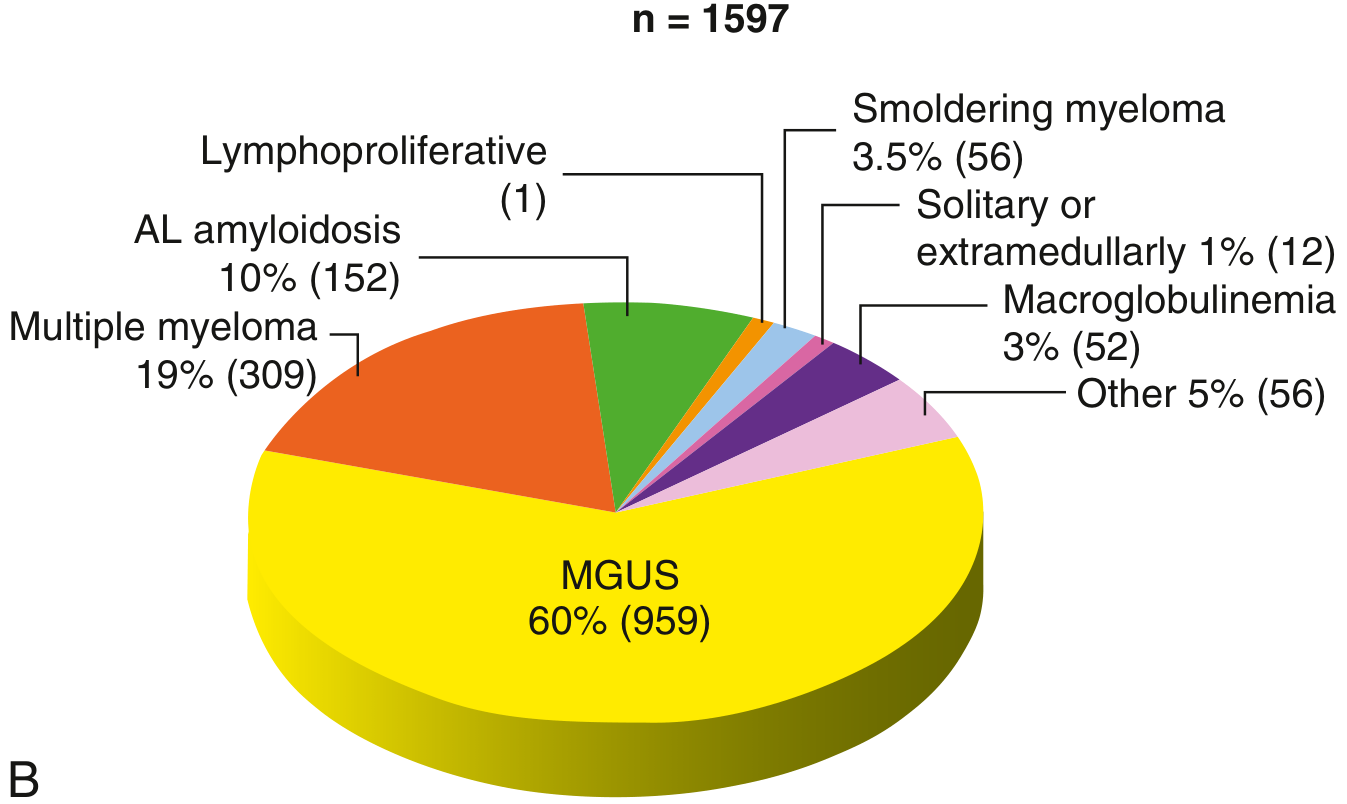
Key takeaway from the chart: Multiple myeloma accounts for only ~19% of M-band cases. The majority (60%) are MGUS.
Full Classification (WHO-5 / Goldman-Cecil)
I. Premalignant Monoclonal Gammopathies (most common overall)
| Entity | Key Features |
|---|---|
| MGUS (IgM type) | M protein <3 g/dL; BM plasma cells <10%; no end-organ damage; risk of progression to Waldenstrom/lymphoma |
| MGUS (Non-IgM type) | Most common MGUS; IgG or IgA; risk of progression to myeloma ~1%/year |
| Light chain MGUS | No intact Ig; only free light chains on urine SPEP/FLC assay |
| Biclonal/triclonal MGUS | Two or three separate M proteins from different clones |
| Smoldering Multiple Myeloma (SMM) | M protein ≥3 g/dL OR BM plasma cells 10-60%; no CRAB criteria; 10% per year progression risk |
| MGRS (Monoclonal Gammopathy of Renal Significance) | Causes renal injury (glomerulonephritis, amyloid) without meeting myeloma criteria |
| Cold Agglutinin Disease (CAD) | IgM M protein causing autoimmune hemolytic anemia |
II. Malignant Monoclonal Gammopathies
| Entity | Key Features |
|---|---|
| Plasma Cell Leukemia | Plasma cells >20% in peripheral blood; aggressive variant of myeloma |
| POEMS Syndrome | Polyneuropathy, Organomegaly, Endocrinopathy, M protein (IgA or IgG lambda), Skin changes; rare |
| Solitary Plasmacytoma | Single lytic lesion, normal BM elsewhere; ~60-70% eventually progress to MM |
| Waldenstrom Macroglobulinemia | IgM M protein >3 g/dL; lymphoplasmacytic lymphoma infiltrating BM; hyperviscosity syndrome |
III. Heavy Chain Diseases (HCDs)
These produce only the heavy chain portion of immunoglobulin (no light chain):
- γ-HCD (Franklin disease) - may mimic lymphoma; palatal edema classic
- α-HCD (Seligmann disease) - most common HCD; malabsorption/diarrhea (Mediterranean lymphoma)
- μ-HCD - resembles CLL; splenomegaly; vacuolated plasma cells
IV. Cryoglobulinemia
- Type I: Monoclonal Ig (usually IgM or IgG) from plasma cell disorder or Waldenstrom
- Type II (Mixed): Monoclonal IgM + polyclonal IgG - most often Hepatitis C associated
- Type III: Polyclonal - connective tissue diseases
V. Immunoglobulin Light Chain (AL) Amyloidosis
- Accounts for ~10% of M-protein cases (chart above)
- Free light chains deposit as amyloid in heart, kidneys, liver, nerves
- Often IgG lambda or free lambda
- Distinguished from MM by organ involvement pattern and absence of CRAB criteria
VI. Other B-cell Lymphoproliferative Disorders That May Produce M Protein
- Chronic Lymphocytic Leukemia (CLL) - small IgM or IgG M band in some cases
- Diffuse Large B-cell Lymphoma (DLBCL)
- Marginal zone lymphoma
- Mantle cell lymphoma
- Follicular lymphoma
VII. Reactive/Secondary M Proteins (Transient or Persistent)
These are not clonal plasma cell disorders but produce a paraprotein-like band:
- Chronic infections: HIV, HCV (Hepatitis C), HBV, tuberculosis, leishmaniasis
- Autoimmune diseases: SLE, RA, Sjogren syndrome
- Liver disease: cirrhosis (beta-gamma bridging can mimic)
- Post organ transplant
- Immunosuppressive therapy
Memory Aid: SPAM-WC
| Letter | Meaning |
|---|---|
| S | Smoldering myeloma |
| P | Plasmacytoma (solitary) / POEMS / Plasma cell leukemia |
| A | AL amyloidosis / Alpha-HCD |
| M | MGUS (IgM, non-IgM, light chain) |
| W | Waldenstrom macroglobulinemia |
| C | CLL / Cryoglobulinemia |
Sources: Goldman-Cecil Medicine, 26e (Table 173-1, Plasma Cell Proliferative Disorders) | Quick Compendium of Clinical Pathology, 5e (WHO-5 classification of monoclonal gammopathies) | Robbins & Kumar Basic Pathology (M protein types in MM)
Tell me differntial of acute lymhoblastic leukemia where tdt is neg and cd3 and cd7 positive
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Excellent - I now have comprehensive textbook data. Let me compile the complete answer.
TdT-negative, CD3+, CD7+ Lymphoid Malignancy: Differential Diagnosis
Why This Immunophenotype Matters
TdT (terminal deoxynucleotidyl transferase) is a marker of immature/precursor lymphoblasts - positive in >95% of ALL cases. A TdT-negative lymphoid malignancy with CD3+ and CD7+ is, by definition, a mature T-cell (post-thymic) neoplasm, NOT a true ALL, and must be re-classified accordingly. This is a critical diagnostic pivot point.
"Mature αβ T cells express CD2, CD3, CD5, CD7, and either CD4 or CD8. They are negative for TdT and CD1." - Henry's Clinical Diagnosis and Management
The Core Differentials
1. T-Cell Prolymphocytic Leukemia (T-PLL) - TOP DIFFERENTIAL
Classic CD3+ CD7-bright, TdT-negative
| Feature | Detail |
|---|---|
| Morphology | Prolymphocytes - medium-sized cells with prominent punched-out nucleolus, cytoplasmic blebs |
| Immunophenotype | CD2+, CD3+ (surface, weak), CD7+ (bright/strong), CD4+ (60%), CD4/CD8 co-expression (25%), CD8+ (15%), TdT-negative |
| Cytogenetics | inv(14)(q11;q32) in 80%, t(14;14) in 10%; trisomy 8q in 70% |
| Presentation | WBC often >100,000/μL, hepatosplenomegaly, lymphadenopathy, skin lesions (20%) |
| Prognosis | Aggressive; median survival ~12 months; responds to alemtuzumab (anti-CD52) |
- Harrison's Principles of Internal Medicine 22e; Henry's Clinical Diagnosis, p. 766
2. T-Cell Large Granular Lymphocytic (LGL) Leukemia
| Feature | Detail |
|---|---|
| Morphology | Large cells with abundant pale blue cytoplasm and azurophilic granules |
| Immunophenotype | CD3+, CD8+, CD57+, TCRαβ, TdT-negative; CD7 variably expressed (may be lost) |
| Presentation | Severe neutropenia ± anemia; mild lymphocytosis (2,000-20,000/μL); splenomegaly; associated with rheumatoid arthritis (Felty-like) |
| Course | Indolent; treatment = cyclosporine, methotrexate, low-dose cyclophosphamide |
3. Adult T-Cell Leukemia/Lymphoma (ATLL)
| Feature | Detail |
|---|---|
| Etiology | HTLV-1 infection; endemic in Japan, Caribbean, Central Africa |
| Immunophenotype | CD3+, CD4+, CD25+ (IL-2 receptor, very characteristic), CD7 often negative or dim; TdT-negative |
| Morphology | "Floret cells" / clover-leaf nuclei |
| Presentation | Hypercalcemia, lytic bone lesions, skin involvement, lymphadenopathy |
| Note | CD7 is typically lost, which helps distinguish from T-PLL |
4. Peripheral T-Cell Lymphoma, NOS (PTCL-NOS)
| Feature | Detail |
|---|---|
| Immunophenotype | CD3+, CD7+/- (CD7 often lost - aberrant loss is diagnostic clue), TdT-negative |
| Presentation | Nodal disease; constitutional B symptoms; older adults |
| Key point | If CD7 is retained (not lost), this supports diagnosis; most PTCLs actually lose CD7 |
| Genetics | Complex karyotypes; no defining translocation |
5. Hepatosplenic T-Cell Lymphoma (HSTL)
| Feature | Detail |
|---|---|
| Immunophenotype | CD3+, CD7+, CD56+, CD4-, CD8-/+, TCRγδ (usually), TdT-negative |
| Presentation | Young adults/adolescents; massive hepatosplenomegaly without lymphadenopathy; associated with immunosuppression (post-transplant, IBD on azathioprine) |
| Prognosis | Very aggressive; poor prognosis |
6. Enteropathy-Associated T-Cell Lymphoma (EATL)
| Feature | Detail |
|---|---|
| Immunophenotype | CD3+, CD7+, CD103+ (gut-homing marker), CD8+/-, TdT-negative |
| Association | Celiac disease; involves jejunum and ileum |
| Presentation | Abdominal pain, perforation, malabsorption |
7. Mycosis Fungoides / Sézary Syndrome
| Feature | Detail |
|---|---|
| Immunophenotype | CD3+, CD4+, CD7 often LOST (CD7 loss is a diagnostic hallmark of MF), TdT-negative |
| Presentation | Cutaneous patches/plaques/tumors (MF); erythroderma + Sézary cells in blood (SS) |
| Note | CD7 negativity is more characteristic here; CD7+ would make SS/MF less likely |
8. Aggressive NK-Cell Leukemia
| Feature | Detail |
|---|---|
| Immunophenotype | Surface CD3- (cytoplasmic CD3+), CD2+, CD56+, CD7 variable; TdT-negative |
| Key distinction | Surface CD3 negative (uses cytoplasmic CD3 polyclonal antibody) - distinguishes from true T-cell neoplasms |
| Association | EBV+, young Asians |
Summary Comparison Table
| Diagnosis | CD3 | CD7 | TdT | CD4/CD8 | Key Distinguishing Feature |
|---|---|---|---|---|---|
| T-PLL | + (weak) | ++ (bright) | - | CD4+ or co-exp | inv(14), very high WBC |
| T-LGL Leukemia | + | +/- | - | CD8+ | Neutropenia, azurophilic granules, RA |
| ATLL | + | - (lost) | - | CD4+, CD25+ | HTLV-1, hypercalcemia, floret cells |
| PTCL-NOS | + | +/- (often lost) | - | Variable | Nodal disease, no defining marker |
| HSTL | + | + | - | CD4-/CD8- | TCRγδ, hepatosplenomegaly, no nodes |
| EATL | + | + | - | CD8+, CD103+ | Celiac disease, GI involvement |
| MF/Sezary | + | usually lost | - | CD4+ | Skin disease, cerebriform cells |
| NK-cell leukemia | surface- (cCD3+) | +/- | - | - | CD56+, EBV+, surface CD3- |
Diagnostic Approach When TdT- CD3+ CD7+
- First confirm: Is CD3 surface or cytoplasmic? (cytoplasmic only = NK-cell)
- Add CD4, CD8, CD56, CD25, CD57, TCRαβ/γδ - narrow the T-cell subtype
- Check CD7 intensity: CD7 bright = T-PLL; CD7 dim/lost = PTCL/ATLL/MF
- HTLV-1 serology if CD4+, CD25+
- EBV if clinical picture suggests NK/T-cell
- TCR gene rearrangement to confirm clonality
- Cytogenetics/FISH: inv(14) = T-PLL; isochromosome 7q = HSTL
Sources: Harrison's Principles of Internal Medicine 22e | Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 765-766 | Robbins & Kumar Pathologic Basis of Disease (Robbins, Cotran)
Tell me all t cell neoplasm which are indolent and aggressive
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
I now have comprehensive, authoritative data from multiple textbooks. Here is the complete answer:
T-Cell (and NK-Cell) Neoplasms: Indolent vs. Aggressive
INDOLENT T-Cell Neoplasms
1. Mycosis Fungoides (MF) - Most Common CTCL
- 5-year survival: 88% (classic MF); Folliculotropic MF: 75%
- Epidermotropic CD4+ T-cell lymphoma; presents as patches → plaques → tumors
- Indolent course over years-decades; may transform to aggressive large-cell lymphoma
- Variants (all indolent):
- Pagetoid reticulosis - 5-year OS ~100%
- Granulomatous slack skin - 5-year OS ~100%
2. Lymphomatoid Papulosis (LyP)
- 5-year survival: 99% - most indolent T-cell disorder
- CD30+ self-regressing papulonodular skin lesions; wax and wane
- Despite histologic appearance of high-grade lymphoma, it is clinically benign
- ~10-20% develop secondary lymphoma (MF, ALCL, Hodgkin)
3. Primary Cutaneous Anaplastic Large Cell Lymphoma (C-ALCL)
- 5-year survival: 95%
- CD30+ solitary/localized skin nodules or tumors; often self-regressing
- ALK-negative (unlike systemic ALCL); excellent prognosis when skin-limited
- Treat with radiation or surgery; chemo only if disseminated
4. T-Cell Large Granular Lymphocytic (LGL) Leukemia
- Indolent; median survival measured in years
- CD3+, CD8+, CD57+, TCRαβ; large granular lymphocytes with azurophilic granules
- Presents with chronic neutropenia, mild lymphocytosis, splenomegaly
- Strongly associated with rheumatoid arthritis (Felty syndrome overlap)
- Treatment: cyclosporine, low-dose methotrexate, cyclophosphamide
5. NK-Cell Chronic Lymphoproliferative Disorder (Indolent NK-LGL Leukemia)
- CD2+, CD56+; no clonal EBV (distinguishes from aggressive NK leukemia)
- Discovered incidentally; no fever, no hepatosplenomegaly, no pancytopenia
- Indolent; may not require treatment
6. Subcutaneous Panniculitis-Like T-Cell Lymphoma (SPTCL) - αβ type
- 5-year survival: ~87%; Indolent
- αβ TCR type = indolent; γδ TCR type = aggressive (now classified separately as PCGD-TCL)
- Subcutaneous nodules mimicking panniculitis; rimming of fat cells
- Responds well to immunosuppression (steroids, cyclosporine)
- Prognosis worsens if hemophagocytic syndrome (HPS) develops
7. Primary Cutaneous CD4+ Small/Medium T-Cell Lymphoproliferative Disorder
- 5-year survival: ~100%
- Usually solitary facial lesion; low risk of dissemination
- Often managed with local excision alone
8. Primary Cutaneous Acral CD8+ T-Cell Lymphoproliferative Disorder
- 5-year survival: 100% (provisional WHO-5 entity - now classed as lymphoproliferative disorder)
- Localized to acral skin; very indolent
AGGRESSIVE T-Cell Neoplasms
1. T-Cell Prolymphocytic Leukemia (T-PLL)
- Median survival: ~12 months - one of the most aggressive T-cell leukemias
- CD2+, CD3+ (weak), CD7++ (bright), CD4+ (60%), TdT-negative
- WBC often >100,000/μL; hepatosplenomegaly, skin involvement
- Cytogenetics: inv(14)(q11;q32) in 80%; trisomy 8q
- Treatment: alemtuzumab (anti-CD52) → allogeneic SCT in remission
2. Adult T-Cell Leukemia/Lymphoma (ATLL)
- Aggressive (acute) form: median survival months; chronic/smoldering forms are more indolent
- HTLV-1 driven; endemic in Japan, Caribbean, Central Africa
- CD3+, CD4+, CD25+ (IL-2 receptor), floret/cloverleaf cells
- Hypercalcemia, lytic lesions, skin involvement, lymphadenopathy
- 4 clinical forms: Acute (most common, most aggressive), Lymphomatous, Chronic (indolent), Smoldering (most indolent)
3. Extranodal NK/T-Cell Lymphoma, Nasal Type
- 5-year survival: 16% overall; as low as 7% with multiple risk factors
- EBV-driven; CD2+, CD56+, cytoplasmic CD3+ (surface CD3-)
- Angioinvasive, angiodestructive; "lethal midline granuloma"
- Common in Asia, Mexico, Central/South America
- Risk factors for poor prognosis: B symptoms, advanced stage, elevated LDH, regional LN involvement
- 5-year OS: 81% (0 risk factors) → 7% (3-4 risk factors)
4. Hepatosplenic T-Cell Lymphoma (HSTL)
- Poor prognosis; median OS ~16 months
- TCRγδ (usually); CD3+, CD7+, CD56+, CD4-, CD8-
- Young adults/adolescents; immunosuppressed (post-transplant, IBD on azathioprine/infliximab)
- Massive hepatosplenomegaly without lymphadenopathy; cytopenias
- Sinusoidal infiltration of liver, spleen, BM
- Isochromosome 7q in most cases
5. Enteropathy-Associated T-Cell Lymphoma (EATL)
- Median survival 7-11 months; very poor prognosis
- Associated with celiac disease (Type I = classic EATL)
- Type II (now: Monomorphic Epitheliotropic Intestinal T-Cell Lymphoma / MEITL) - not celiac-related; worse prognosis
- Presents with abdominal pain, perforation, malabsorption
- CD3+, CD7+, CD103+, CD8+; TIA-1+
6. Peripheral T-Cell Lymphoma, NOS (PTCL-NOS)
- 5-year survival: 15-30%; aggressive
- Diagnosis of exclusion after ruling out specific subtypes
- CD3+, usually CD4+; loss of CD5/CD7 = worse prognosis
- B symptoms, generalized lymphadenopathy, advanced stage at presentation
- Treatment: CHOP or CHOEP; brentuximab-CHP if CD30+
7. Angioimmunoblastic T-Cell Lymphoma (AITL) / Nodal TFH Lymphoma
- Median OS: 15-36 months; aggressive
- CD4+ TFH-derived: CD3+, CD10+, CXCL13+, PD-1+, BCL6+
- Systemic disease: fever, rash, polyarthritis, hemolytic anemia, polyclonal hypergammaglobulinemia
- EBV+ immunoblasts (risk of secondary EBV+ B-cell lymphoma)
- Mutations: TET2 (76%), DNMT3 (33%), IDH2 (20%)
8. Anaplastic Large Cell Lymphoma (ALCL)
- Two distinct entities with different prognosis:
| Type | 8-year OS | Key Feature |
|---|---|---|
| ALK-positive ALCL | 82% - relatively favorable | t(2;5); younger patients |
| ALK-negative ALCL | 49% - aggressive | Older; DUSP22-R subset has better prognosis |
| Breast implant-associated ALCL (BIA-ALCL) | Excellent (~indolent) | ALK-negative; localized to capsule |
| Primary cutaneous ALCL (C-ALCL) | 95% - indolent | Skin-limited; self-regressing |
- "Hallmark cells": horseshoe-shaped nuclei, prominent nucleoli; CD30++ (all types)
- Treatment: CHOP/CHOEP; Brentuximab vedotin (anti-CD30) for relapsed; crizotinib for relapsed ALK+ ALCL
9. Aggressive NK-Cell Leukemia
- Rapidly progressive; median survival weeks-months
- CD2+, CD56+, cytoplasmic CD3+; clonal EBV+
- Young Asians; fever, pancytopenia, hepatosplenomegaly, HPS, multiorgan failure
- del(6), del(17); serum Fas ligand elevated
10. Primary Cutaneous Gamma/Delta T-Cell Lymphoma (PCGD-TCL)
- 5-year survival: 11% - one of the most aggressive CTCLs
- TCRγδ; CD3+, CD56+, cytotoxic markers; CD4-, CD8-
- Disseminated skin lesions with mucosal/visceral involvement
- Formerly confused with SPTCL (now separated)
11. Primary Cutaneous Aggressive Epidermotropic CD8+ Cytotoxic T-Cell Lymphoma
- 5-year survival: 31% - aggressive
- Widespread skin lesions with epidermotropism; may disseminate to viscera
- CD8+, TIA-1+, granzyme B+; CD4-
Summary Table
| Category | Entity | 5-yr OS | Course |
|---|---|---|---|
| INDOLENT | Mycosis Fungoides (classic) | 88% | Indolent |
| Lymphomatoid Papulosis | 99% | Very indolent | |
| Primary Cutaneous ALCL (C-ALCL) | 95% | Indolent | |
| T-LGL Leukemia | Years | Indolent | |
| NK-LGL Chronic (no EBV) | Years | Indolent | |
| SPTCL (αβ type) | 87% | Indolent | |
| ATLL - Chronic/Smoldering | Variable | Indolent | |
| ALK+ ALCL (systemic) | 82% (8-yr) | Intermediate | |
| BIA-ALCL | Excellent | Indolent | |
| AGGRESSIVE | T-PLL | <20% (1 yr) | Very aggressive |
| Aggressive NK-cell leukemia | <10% | Very aggressive | |
| PCGD-TCL | 11% | Very aggressive | |
| Extranodal NK/T-cell (nasal) | 16% avg | Aggressive | |
| EATL / MEITL | ~20% | Very aggressive | |
| HSTL | Poor | Very aggressive | |
| PTCL-NOS | 15-30% | Aggressive | |
| AITL / Nodal TFH | 30-40% | Aggressive | |
| ALK-negative ALCL | 49% | Aggressive | |
| ATLL (acute/lymphomatous) | Poor | Very aggressive | |
| Cutaneous aggressive CD8+ CTCL | 31% | Aggressive | |
| SPTCL (γδ type / PCGD-TCL) | 11% | Aggressive |
Sources: Dermatology 2-Volume Set 5e (WHO-EORTC Classification Table 120.1) | Harrison's Principles of Internal Medicine 22e | Henry's Clinical Diagnosis and Management by Laboratory Methods (Table 34.7) | Quick Compendium of Clinical Pathology 5e (WHO-5 / ICC classification)
Also do the same for b cell neoplasm
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
I have excellent comprehensive data from Harrison's and Tietz. The large truncated result contains extensive B-cell lymphoma data. Let me compile the complete answer now:
B-Cell Neoplasms: Indolent vs. Aggressive
INDOLENT B-Cell Neoplasms
1. Chronic Lymphocytic Leukemia / Small Lymphocytic Lymphoma (CLL/SLL) - Most Common Adult Leukemia
- Course: Indolent; median survival 7-10+ years (highly variable by stage)
- Immunophenotype: CD5+, CD19+, CD20 (dim), CD23+, CD10-, cyclin D1-; surface Ig dim
- Genetics: del(13q) = best prognosis; del(11q), del(17p)/TP53 = poor prognosis
- Mutation: IGHV mutated = better prognosis; IGHV unmutated = worse
- Key point: CLL (leukemic) and SLL (nodal) are the same disease; Richter transformation to DLBCL = aggressive conversion (~5-10%)
- Treatment (when needed): BTK inhibitors (ibrutinib, acalabrutinib), BCL-2 inhibitor (venetoclax), anti-CD20 (rituximab/obinutuzumab)
- Cytogenetics: del(13q14) most common; trisomy 12; del(11q); del(17p) - worst
2. Follicular Lymphoma (FL) - Most Common Indolent Lymphoma
- Course: Indolent; median survival 8-15 years; Grade 1-2 = indolent; Grade 3B = aggressive (treat like DLBCL)
- Immunophenotype: CD10+, CD19+, CD20+, BCL2+, BCL6+, CD5-, CD23-
- Genetics: t(14;18) BCL2/IgH - hallmark translocation (85-90%); BCL2 overexpression prevents apoptosis
- Key features: Follicular architecture; "watch and wait" often appropriate for asymptomatic low-burden disease
- Transformation: ~30% transform to DLBCL over lifetime (histologic transformation = poor prognosis)
- Treatment: Rituximab ± chemotherapy (R-CHOP, R-bendamustine); lenalidomide; PI3K inhibitors for relapsed
3. Marginal Zone Lymphomas (MZL) - Three Subtypes, All Indolent
| Subtype | Location | Key Association | Genetics | Notes |
|---|---|---|---|---|
| Extranodal MZL (MALT lymphoma) | GI (stomach #1), lung, salivary, thyroid | H. pylori (gastric); Sjogren (salivary); Hashimoto (thyroid) | t(11;18) API2/MALT1 | Gastric MALT may regress with H. pylori eradication alone |
| Splenic MZL | Spleen, blood, BM | Hepatitis C | Trisomy 3; del(7q) | Villous lymphocytes in blood; splenectomy often curative |
| Nodal MZL | Lymph nodes | Least common MZL | Complex | Diagnosis of exclusion |
- Immunophenotype (all): CD19+, CD20+, CD5-, CD10-, CD23-, BCL2+, surface Ig bright
4. Hairy Cell Leukemia (HCL)
- Course: Indolent; very good prognosis with treatment (CR rates ~90%)
- Immunophenotype: CD19+, CD20+ (bright), CD11c+, CD25+, CD103+, CD123+, CD200+, Annexin A1+ (specific marker); cyclin D1+ (weak)
- Genetics: BRAF V600E mutation in ~100% of classic HCL (absent in HCL-variant and other B-cell LPDs)
- Morphology: "Hairy" cytoplasmic projections; TRAP (tartrate-resistant acid phosphatase) positive
- Presentation: Pancytopenia, massive splenomegaly, monocytopenia, "dry tap" on BM biopsy
- Treatment: Purine analogues (cladribine, pentostatin) = highly effective; BRAF inhibitor (vemurafenib) for refractory cases
5. Lymphoplasmacytic Lymphoma (LPL) / Waldenstrom Macroglobulinemia (WM)
- Course: Indolent; median survival 5-10 years
- Hallmark: IgM monoclonal protein + hyperviscosity syndrome (headache, visual changes, bleeding, Raynaud's)
- Mutation: MYD88 L265P mutation in ~90% - key diagnostic mutation; CXCR4 mutations in ~30%
- Immunophenotype: CD19+, CD20+, CD25+, CD138 (plasma cell component), surface IgM+, CD5-, CD10-
- Key features: Lymphoplasmacytic morphology; BM infiltration; hyperviscosity requires urgent plasmapheresis
- Treatment: BTK inhibitors (ibrutinib), rituximab ± chemotherapy; plasmapheresis for hyperviscosity
6. Splenic Diffuse Red Pulp Small B-Cell Lymphoma
- Course: Indolent; rare entity
- Often BRAF V600E negative (distinguishes from HCL); villous lymphocytes
7. Nodular Lymphocyte-Predominant Hodgkin Lymphoma (NLPHL) (B-cell origin)
- Course: Indolent; excellent prognosis (>90% 10-year OS)
- CD20+, CD45+, CD79a+; CD15-, CD30- (distinguishes from classic HL)
- "Popcorn cells" / LP cells; CD4+ T-cell rosetting
- ~5% transform to DLBCL (usually LP cell transformation)
8. Primary Cutaneous Marginal Zone Lymphoma
- 5-year OS: ~99% - extremely indolent
- Skin-limited; rarely disseminates
- Associated with Borrelia burgdorferi in some European cases
9. Primary Cutaneous Follicle Centre Lymphoma
- 5-year OS: ~95% - indolent
- CD10+, BCL6+, BCL2 usually negative (unlike nodal FL)
- Localized skin lesions; radiation often curative
AGGRESSIVE B-Cell Neoplasms
1. Diffuse Large B-Cell Lymphoma (DLBCL) - Most Common Aggressive Lymphoma
- Course: Aggressive; but ~60-70% curable with R-CHOP
- Immunophenotype: CD19+, CD20+, CD79a+; variable CD10, BCL6, IRF4/MUM1
- Molecular subtypes (cell of origin):
- GCB (Germinal Center B-cell): CD10+, BCL6+; t(14;18) BCL2; better prognosis
- ABC/Non-GCB (Activated B-cell): IRF4/MUM1+; NF-κB activation; CARD11, MYD88 mutations; worse prognosis
- Special subtypes:
- Double-hit (DHL): MYC + BCL2 rearrangements - very aggressive
- Triple-hit (THL): MYC + BCL2 + BCL6 - worst prognosis
- Double-expressor: Overexpression of MYC + BCL2 protein (without rearrangement)
- Primary DLBCL of CNS: CD10-, BCL6+, MUM1+; treated with HD-MTX-based regimens
- Primary mediastinal (thymic) large B-cell lymphoma (PMBCL): young women; CD30+, CD23+; distinct from DLBCL
- EBV+ DLBCL: Older patients; aggressive
- Primary testicular DLBCL: High CNS relapse risk; requires CNS prophylaxis
- Treatment: R-CHOP; CAR-T (axicabtagene, tisagenlecleucel) for relapsed/refractory
2. Mantle Cell Lymphoma (MCL)
- Course: Aggressive but generally incurable; median survival 3-7 years
- The exception: Indolent/leukemic non-nodal MCL = truly indolent subset (SOX11-negative)
- Hallmark: t(11;14) BCL1/IgH → cyclin D1 overexpression (virtually diagnostic)
- Immunophenotype: CD5+, CD19+, CD20+, cyclin D1+, SOX11+, CD10-, CD23-
- "Blastoid variant" = most aggressive form
- High risk for GI involvement (lymphomatous polyposis)
- Treatment: Aggressive regimens (R-CHOP/R-DHAP alternating); BTK inhibitors (ibrutinib) for relapsed; auto-SCT consolidation
3. Burkitt Lymphoma (BL)
- Course: Extremely aggressive; but highly curable with intensive chemotherapy (~80-90% cure in children)
- The fastest-growing human tumor - doubling time ~24-48 hours; Ki-67 ~100%
- Genetics: t(8;14) MYC/IgH (80%); t(2;8) or t(8;22) - ALL involve MYC
- Immunophenotype: CD10+, CD19+, CD20+, BCL6+, BCL2- (key!), TdT-, Ki-67 ~100%
- 3 clinical variants:
- Endemic (African): EBV-driven; jaw/facial bones; children
- Sporadic: Ileocecal mass; abdominal; children/young adults; EBV in ~30%
- Immunodeficiency-associated: HIV; EBV in ~40%
- "Starry sky" appearance on histology (macrophages ingesting apoptotic tumor cells)
- Treatment: Intensive short-duration chemotherapy (R-CODOX-M/IVAC, R-hyperCVAD); tumor lysis syndrome prophylaxis mandatory
4. High-Grade B-Cell Lymphoma with MYC and BCL2/BCL6 Rearrangements (Double/Triple Hit)
- Course: Very aggressive; 2-year OS ~30-50%
- Formally separated from DLBCL in WHO classification
- Often presents as de novo or as transformation of FL
- Treatment: More intensive than R-CHOP required (DA-EPOCH-R preferred); early auto-SCT
5. Primary Mediastinal (Thymic) Large B-Cell Lymphoma (PMBCL)
- Course: Aggressive but potentially curable (~80% long-term survival)
- Young women; bulky anterior mediastinal mass; SVC syndrome
- CD30+, CD23+, MHC class II loss; CD10-, surface Ig-
- Genetically similar to nodular sclerosis Hodgkin lymphoma
- Treatment: DA-EPOCH-R (dose-adjusted); avoids routine radiation if PET-negative at end of therapy
6. Plasmablastic Lymphoma
- Course: Very aggressive; poor prognosis (median OS ~15 months)
- Associated with HIV/immunosuppression; EBV+
- Plasma cell differentiation: CD138+, CD38+, MUM1+; CD20- (loses B-cell markers)
- High MYC expression; Ki-67 very high
- Involves jaw, oral cavity, GI tract
7. Primary Effusion Lymphoma (PEL)
- Course: Very aggressive; median OS ~6 months
- HHV-8 (KSHV) driven; EBV co-infection common
- HIV/immunosuppressed patients
- Presents as lymphomatous effusion (pleural, pericardial, peritoneal) WITHOUT a tumor mass
- CD19-, CD20- (loses B-cell markers); CD138+, CD38+, HHV-8+
8. Intravascular Large B-Cell Lymphoma
- Course: Aggressive; often fatal if unrecognized
- Tumor cells confined to lumens of small vessels; no tumor mass
- Presents with CNS/neurological symptoms, skin lesions, fever, cytopenias
- Random skin biopsy may be diagnostic
9. CNS B-Cell Lymphoma (Primary CNS Lymphoma - PCNSL)
- Course: Aggressive; median OS 2-5 years with treatment
- Almost always DLBCL histology; CD10-, BCL6+, MUM1+ (ABC subtype)
- Periventricular lesions; steroid "melting sign"
- Treatment: High-dose methotrexate-based regimens; avoid whole-brain radiation if possible (neurotoxicity)
Summary Classification Table
INDOLENT
| Entity | Key Marker/Genetics | 5-yr OS | Hallmark |
|---|---|---|---|
| CLL/SLL | CD5+, CD23+, CD20(dim) | 70-80%+ | Most common adult leukemia |
| Follicular Lymphoma (Gr 1-2) | BCL2+, t(14;18), CD10+ | 75-85% | BCL2/IgH translocation |
| Gastric MALT | CD5-, CD10- | >90% | H. pylori; t(11;18) |
| Splenic MZL | Villous lymphocytes | >85% | Hep C; del(7q) |
| Hairy Cell Leukemia | CD103+, BRAF V600E, Annexin A1 | >95% | TRAP+; "dry tap" |
| LPL / Waldenstrom | MYD88 L265P, IgM M-band | 60-70% (5yr) | Hyperviscosity; MYD88 L265P |
| Primary Cutaneous FL | CD10+, BCL2- | ~95% | Skin-limited |
| Primary Cutaneous MZL | CD5-, CD10- | ~99% | Borrelia; skin-only |
| NLPHL | CD20+, CD30-, CD15- | >90% | Popcorn cells |
AGGRESSIVE
| Entity | Key Marker/Genetics | Prognosis | Hallmark |
|---|---|---|---|
| DLBCL (GCB) | CD10+, BCL6+, t(14;18) | 60-70% curable | R-CHOP curative |
| DLBCL (ABC/non-GCB) | MUM1+, MYD88 mut | Worse than GCB | NF-κB activation |
| Double/Triple Hit Lymphoma | MYC + BCL2 ± BCL6 | ~30-50% (2yr) | Most aggressive DLBCL |
| Mantle Cell Lymphoma | Cyclin D1+, t(11;14), CD5+ | 3-7 yr median | BCL1/IgH; GI polyposis |
| Burkitt Lymphoma | MYC t(8;14), BCL2-, Ki-67~100% | 80-90% (children) | Fastest growing; starry sky |
| PMBCL | CD30+, CD23+, mediastinal | ~80% curable | Young women; mediastinum |
| Plasmablastic Lymphoma | CD138+, CD20-, EBV+, HIV | Poor (~15 mo) | HIV; jaw; no CD20 |
| Primary Effusion Lymphoma | HHV-8+, CD20-, effusions | Very poor (~6 mo) | Body cavity; no mass |
| CNS Lymphoma (PCNSL) | ABC-DLBCL; BCL6+, MUM1+ | 2-5 yr | HD-MTX required |
| Intravascular LBCL | CD20+; intravascular | Poor if unrecognized | Skin/CNS; no mass |
Important "Middle Ground" - Context-Dependent Behavior
| Entity | Why It's In-Between |
|---|---|
| Follicular Lymphoma Grade 3B | Treat aggressively like DLBCL |
| MCL (leukemic/non-nodal, SOX11-) | Truly indolent subset; watch-and-wait |
| CLL → Richter transformation | Switches from indolent to aggressive (DLBCL) |
| FL → DLBCL transformation | ~30% lifetime risk |
| ATLL chronic/smoldering | Indolent forms of an otherwise aggressive disease |
Sources: Harrison's Principles of Internal Medicine 22e (Lymphoma Chapter - genetics table, B-cell NHL sections) | Tietz Textbook of Laboratory Medicine 7e (Mature B-Cell Lymphoproliferative Disorders) | Henry's Clinical Diagnosis and Management by Laboratory Methods (Table 34.7)
Plasmablastic lymphoma all details including trials
Finding Sources
Searching PubMed
plasmablastic lymphoma treatment outcomes
Reading File
Reading File
Reading Article
PMID: 42138287
Reading Article
PMID: 40167854
Reading Article
PMID: 41347973
Reading Article
PMID: 37453866
Reading Article
PMID: 35961783
Searching PubMed
plasmablastic lymphoma bortezomib daratumumab BCMA CAR-T
Reading Article
PMID: 38767403
Searching PubMed
plasmablastic lymphoma daratumumab bortezomib EPOCH
Searching the Web
plasmablastic lymphoma 2024 2025 clinical trials daratumumab BCMA treatment outcomes
Reading Article
PMID: 38468838
Searching PubMed
plasmablastic lymphoma BCMA bispecific antibody CAR-T autologous transplant
Your credits are exhausted. You can purchase a top-up or subscribe to a plan to continue using Orris. If you're already subscribed, your daily credits will reset tomorrow.
Plz do it
Your credits are exhausted. You can purchase a top-up or subscribe to a plan to continue using Orris. If you're already subscribed, your daily credits will reset tomorrow.
This is a shared conversation. Sign in to Orris to start your own chat.