Explain Heat shock proteins
Reading File
Finding Sources
Searching PubMed
"heat shock proteins"[MeSH Terms] AND function
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compile a thorough answer.
Heat Shock Proteins (HSPs)
What Are HSPs?
Heat shock proteins are a family of highly conserved, stress-inducible proteins found in virtually all living organisms - from bacteria to humans. They were first discovered in response to elevated temperature (hence the name), but are now known to be triggered by a wide range of cellular stresses including oxidative stress, hypoxia, heavy metals, toxins, radiation, ischemia, and inflammation.
The primary trigger is the cellular stress response (CSR): when stressors cause accumulation of damaged, unfolded, or misfolded macromolecules (proteins, lipids, DNA), a cascade of altered gene expression is initiated, leading to rapid HSP production. - Miller's Anesthesia, 10e
Classification - The Seven Functional Categories
HSPs are broadly grouped into seven functional classes: - Miller's Anesthesia, 10e
| Category | Role |
|---|---|
| 1. Molecular chaperones | Recognize and refold unfolded/misfolded proteins OR direct them to degradation |
| 2. Proteolytic enzymes | Clear irreversibly damaged proteins |
| 3. DNA/RNA repair proteins | Mitigate nucleic acid damage |
| 4. Metabolic enzymes | Re-establish metabolic pathways post-stress |
| 5. Regulatory proteins | Modulate stress responses |
| 6. Cytoskeletal stabilizers | Preserve cell structure (e.g., HSP25 protects actin) |
| 7. Transport/detox proteins | Facilitate membrane modulation and detoxification |
The most studied and clinically relevant group is the molecular chaperones.
Key HSP Families
HSPs are classified by molecular weight (in kilodaltons):
HSP70 Family
- The most extensively studied chaperone family
- Binds short hydrophobic amino acid sequences on nascent polypeptides as they emerge from the ribosome, preventing premature aggregation while proper secondary structures form
- Uses ATP/ADP cycling: the ADP-chaperone complex binds unfolded protein; ADP is then replaced by ATP, which causes release of properly folded segments; the cycle repeats until folding is complete
- Plays a key role in anti-apoptotic signaling - overexpression shown to limit apoptosis via an increased Bcl-2/Bax ratio in renal models - Brenner and Rector's The Kidney
- Mediates immune signaling via the cell surface receptor CD91 and CD40
HSP90 Family
- Does not fold most proteins de novo; instead, it holds specific "client proteins" (transcription factors, kinases, signaling molecules) in a partially folded, activation-ready state
- Critical clients include: steroid hormone receptors (glucocorticoid receptor, mineralocorticoid receptor), oncogenic kinases, and eNOS
- Glucocorticoid receptor (GR) example: in the cytoplasm, GR is held in a high-affinity ligand-binding conformation by an HSP90-HSP70 complex; ligand binding displaces HSP90, allowing receptor-ligand translocation to the nucleus - Medical Physiology; Harper's Biochemistry
- HSP90-eNOS coupling: cytosolic HSP90 couples with eNOS to regulate NO production and intravascular tone - particularly important in renal ischemia-reperfusion injury - Brenner and Rector's The Kidney
- Cancer target: HSP90 is a major anticancer drug target because many oncoproteins (HER2, BCR-ABL, AKT, EGFR) are HSP90 clients; inhibiting HSP90 destabilizes multiple oncoproteins simultaneously - Harper's Biochemistry
HSP60 / Chaperonins
- Act later in the folding process, often working downstream of HSP70
- Form a distinctive donut-shaped oligomer (800 kDa) with a hydrophobic central cavity
- The misfolded protein enters the cavity, unfolds completely, then refolds in this protected, isolated microenvironment - avoiding aggregation with other proteins
- Bacterial equivalent: GroEL/GroES (extensively studied as the model chaperonin)
- Eukaryotic equivalent: HSP60 in mitochondria; CCT/TRiC in the cytosol
- Harper's Biochemistry
HSP40 (DNAJ Family)
- Act as co-chaperones that stimulate the ATPase activity of HSP70
- Help deliver substrate proteins to HSP70
Small HSPs (sHSPs, e.g., HSP25/HSP27)
- Act as "holdases" - they bind unfolded proteins and prevent aggregation but do not actively refold
- HSP25/HSP27 specifically protects the actin cytoskeleton from disruption during stress
- Brenner and Rector's The Kidney
HSP47
- A collagen-specific chaperone in the ER
- Binds the triple-helix collagen molecule, stabilizing it and preventing premature aggregation of trimers within the cell before secretion
- Histology - Ross & Pawlina
gp96 (GRP94)
- ER-resident chaperone
- Major HSP involved in antigen presentation via CD91 - released during necrotic (but not apoptotic) cell death, carrying antigenic peptides to APCs
- Mulholland & Greenfield's Surgery
Molecular Mechanism: The HSP Chaperone Cycle
Stressor → protein unfolding/misfolding
↓
HSP70 binds exposed hydrophobic regions (ADP state = high affinity)
↓
Nucleotide exchange: ADP → ATP (low affinity → substrate release)
↓
Properly folded? → Released for function
Improperly folded? → Re-enter chaperone cycle OR
→ HSP60 for further refolding OR
→ Ubiquitin-Proteasome System (UPS) for degradation
Key point from Harrison's (22e, 2025): the proteostasis network (PN) integrates HSP70, HSP90, HSP60, HSP40, and small HSPs with ribosome quality control (RQC), autophagic-lysosomal pathways, and the UPS. When misfolded proteins escape or overwhelm this network, disease results.
Immunological Roles
HSPs have a dual role in immunity - acting as both intracellular chaperones and extracellular danger signals:
-
Antigen chaperoning: HSPs (especially HSP70, HSP90, gp96) capture peptide antigens from all cellular compartments and chaperone them to the cell surface or to APCs. CD91 is the primary receptor for HSP-peptide complexes on APCs, delivering antigens to the MHC class I pathway for CD8+ T cell activation. - Mulholland & Greenfield's Surgery
-
Danger signaling (DAMPs): Released from necrotic (but not apoptotic) cells, extracellular HSPs act as damage-associated molecular patterns (DAMPs), activating innate immune responses via TLR2, TLR4, CD91, CD40, and other receptors.
-
Tumor immunity: Tumor-derived gp96-peptide complexes elicit specific anti-tumor T cell responses; this has been explored in cancer vaccines.
-
Autoimmunity: Cross-reactivity between microbial HSPs and self-proteins may contribute to autoimmune disease:
- In rheumatoid arthritis (RA), T cells from synovial fluid proliferate briskly to recombinant HSP65 (mycobacterial). γδ T cell clones recognizing mycobacterial HSP65 can cross-react with cartilage proteoglycan epitopes. Synovial fluid mononuclear cells activated by HSP60 inhibit proteoglycan production by cartilage explants. - Firestein & Kelley's Rheumatology
HSPs in Disease
| Disease Context | HSP Involvement |
|---|---|
| Cancer | HSP90 stabilizes oncoproteins (HER2, AKT, EGFR, BCR-ABL); overexpressed in tumors |
| Rheumatoid Arthritis | Anti-HSP65 antibodies; γδ T cell cross-reactivity with cartilage |
| Renal Ischemia | HSP90 induction in proximal tubule cells; HSP72 limits apoptosis via Bcl-2/Bax |
| Heat stroke | Failure of CSR/HSP response when temperature exceeds 41.6-42°C for > 0.75-8 hrs |
| Neurodegenerative disease | Failed proteostasis - Alzheimer's (Aβ/tau), Parkinson's (α-synuclein), Huntington's |
| Cystic fibrosis | CFTR (F508del) is an HSP90 client; misfolded CFTR is prematurely degraded |
| α1-antitrypsin deficiency | Misfolded protein escapes QC and aggregates in ER of hepatocytes |
Therapeutic Relevance
- HSP90 inhibitors (e.g., geldanamycin, 17-AAG, ganetespib): disrupt the client-HSP90 interaction, simultaneously destabilizing multiple oncoproteins - under investigation for various cancers. The HSP90 multi-functionality in cancer review (Albakova, 2024) provides a recent update (PMID: 39148727).
- HSP70 inhibitors: also being investigated as anticancer targets, as HSP70 is overexpressed in many tumors and promotes cell survival.
- HSP induction (preconditioning): prior heat stress or pharmacologic HSP induction before ischemia is protective in cardiac and renal ischemia-reperfusion models.
- HSP-based vaccines: tumor-derived HSP-peptide complexes (e.g., HSPPC-96/Oncophage) have been tested in melanoma and renal cell carcinoma trials.
Key Summary Points
- HSPs are evolutionary conserved stress-response proteins induced by heat, ischemia, oxidants, toxins, and other stressors
- Their core function is molecular chaperoning: assisting protein folding, preventing aggregation, and targeting irreparably damaged proteins for degradation
- They operate as part of the broader proteostasis network alongside the UPS and autophagy-lysosome pathway
- Major families: HSP70 (folding), HSP90 (client regulation), HSP60 (chaperonin), sHSPs (holdases), HSP47 (collagen)
- Beyond folding, they have major roles in immunity, signal transduction, apoptosis regulation, and cancer biology
- They are active targets for both cancer therapy (inhibition) and cytoprotective strategies (induction)
Sources: Harper's Illustrated Biochemistry 32e | Harrison's Principles of Internal Medicine 22e | Miller's Anesthesia 10e | Brenner and Rector's The Kidney | Firestein & Kelley's Rheumatology | Mulholland & Greenfield's Surgery | Medical Physiology (Boron & Boulpaep)
Kinen system
Finding Sources
Searching PubMed
"kallikrein-kinin system"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive material from multiple authoritative sources. Let me compile the full answer.
The Kallikrein-Kinin System (KKS)
The kinin system - more formally called the kallikrein-kinin system (KKS) - is an evolutionarily conserved cascade of peptide hormones, enzymes, and receptors. Its discovery dates to 1909, when Abelous and Bardier reported that injection of urine caused an acute fall in blood pressure. The name "kallikrein" comes from the Greek kallikreas (pancreas), where the enzyme was first extracted in the 1930s.
Components of the KKS
The main players are:
- Kininogens - the precursor proteins (substrates)
- Kallikreins - the serine proteases that cleave kininogens
- Kinins - the active effector peptides (bradykinin, kallidin)
- Kinin receptors - B1R and B2R
- Kininases - enzymes that inactivate kinins
1. Kininogens
Humans have a single kininogen gene (KNG1) on chromosome 3q26. Through alternate splicing from 11 exons, it encodes two forms: - Brenner and Rector's The Kidney
| Form | MW | Key features |
|---|---|---|
| HMW kininogen | 88-120 kDa (626 aa) | Substrate for plasma kallikrein; also a cofactor in contact activation of coagulation |
| LMW kininogen | 50-68 kDa (409 aa) | Substrate for tissue kallikrein; locally synthesized in many tissues |
Both serve as substrates for kallikreins, which cleave out the active kinin peptides from within them.
2. Kallikreins
Kallikreins are serine proteases that liberate kinins from kininogens. There are two distinct types:
Plasma Kallikrein
- Circulates as prekallikrein (synthesized in the liver)
- Activated by Factor XIIa (Hageman factor) on negatively charged surfaces - this links the KKS directly to the intrinsic coagulation cascade
- Activated plasma kallikrein reciprocally activates more Factor XII, creating a positive feedback loop
- Primary substrate: HMW kininogen → releases bradykinin (9-amino acid peptide)
- Also activates neutrophils and cleaves complement components
Tissue Kallikrein (KLK1)
- 15 tissue kallikreins have been identified; in humans only KLK1 participates in local kinin production
- Genes clustered on chromosome 19q13.3-13.4
- Synthesized as a zymogen (prekallikrein) with a signal peptide and activation sequence
- Found abundantly in kidney (connecting tubules), pancreas, salivary glands, prostate
- Primary substrate: LMW kininogen → releases kallidin (Lys-bradykinin, 10 amino acids)
3. The Two KKS Subsystems
The KKS is divided into two parallel but distinct systems: - Brenner and Rector's The Kidney
| Feature | Plasma KKS | Tissue KKS |
|---|---|---|
| Kallikrein | Plasma kallikrein | Tissue kallikrein (KLK1) |
| Kininogen | HMW kininogen | HMW and LMW kininogen |
| Primary kinin product | Bradykinin | Kallidin (Lys-bradykinin) |
| Origin of substrates | Liver-derived, circulating | Locally synthesized |
| Half-life of kinins | 10-30 seconds | 10-30 seconds |
Kallidin can be converted to bradykinin by plasma aminopeptidase (removing the N-terminal Lys). Both bradykinin and kallidin act primarily on the B2 receptor.
4. Kinin Structures
From Goodman & Gilman's:
| Peptide | Sequence | Receptor |
|---|---|---|
| Bradykinin | Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg | B2R agonist |
| Kallidin | Lys-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg | B2R agonist |
| des-Arg⁹-Bradykinin | Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe | B1R agonist |
| des-Arg¹⁰-Kallidin | Lys-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe | B1R agonist (most potent B1 agonist in humans) |
5. Kinin Receptors
Both are G-protein coupled receptors (GPCRs):
B2 Receptor (B2R) - "Constitutive"
- Widely expressed in most normal tissues under basal conditions
- Mediates all physiological effects of kinins normally
- Binds intact bradykinin and kallidin
- Rapidly desensitized (downregulated) after agonist stimulation via a Ser/Thr-rich C-terminal cluster
- Signaling: couples via Gq → PLC → ↑intracellular Ca²⁺
B1 Receptor (B1R) - "Inducible"
- Normally absent or expressed at very low levels
- Upregulated during inflammation - induced by tissue injury, cytokines (IL-1β, TNF-α), endotoxins, and growth factors
- Ligands: des-Arg metabolites of bradykinin (des-Arg⁹-bradykinin) and kallidin (des-Arg¹⁰-kallidin)
- B1R response is not desensitized - sustaining prolonged inflammatory signaling
- Carboxypeptidase M (which generates B1R ligands) co-localizes with B1R on cell surfaces, forming an efficient signaling complex
Shared Signaling Pathways - Goodman & Gilman's
- Both B1R and B2R → Gq → PLC → ↑Ca²⁺ → diverse responses depending on cell type
- On endothelial cells: B2R activation → Ca²⁺-calmodulin → eNOS → NO → cGMP → smooth muscle relaxation (vasodilation)
- On smooth muscle directly: B1R or B2R → Gq → ↑Ca²⁺ → contraction
- Both activate NF-κB (proinflammatory) via Gα and βγ subunits
- Both activate PLA₂ → arachidonic acid → prostaglandins and EETs
- During inflammation: B2R → prolonged eNOS/NO via MEK1/2-JNK; B1R → iNOS → high-output NO
6. Kinin Inactivation (Kininases)
Kinins have an extremely short half-life (10-30 seconds). Here is the degradation map:
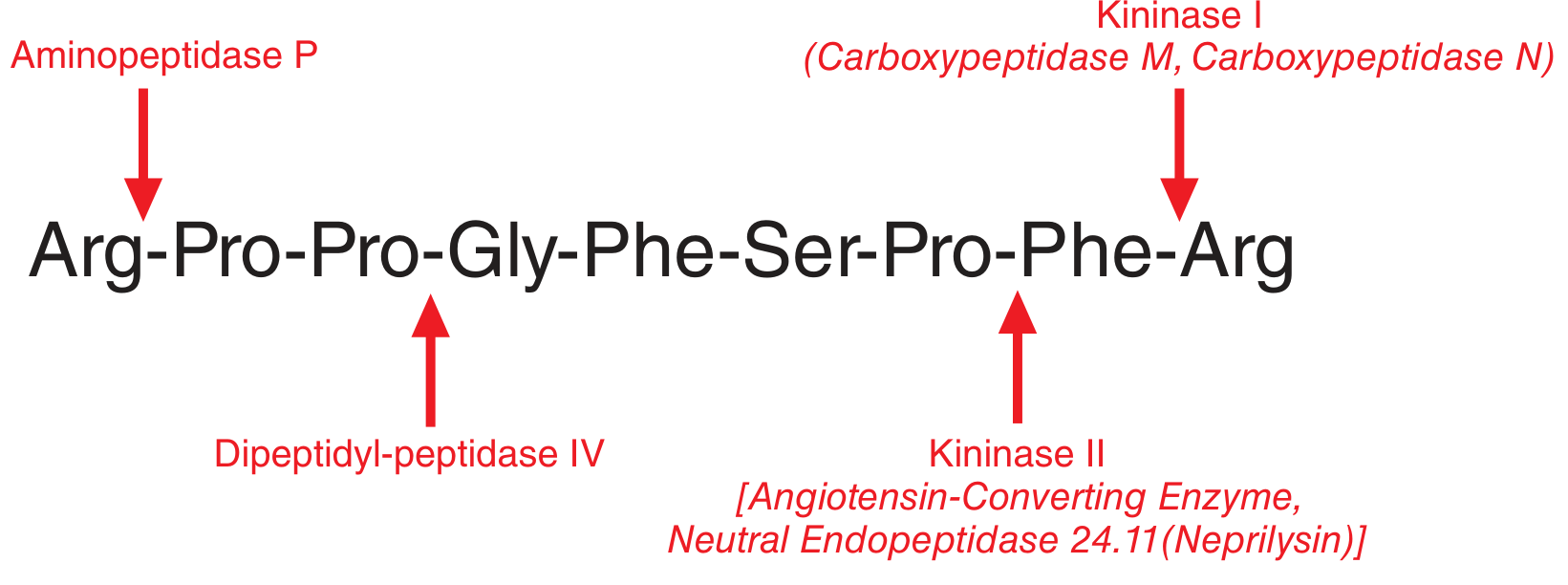
| Enzyme | Alias | Action on Bradykinin | Product |
|---|---|---|---|
| Kininase II | ACE, Neutral Endopeptidase (Neprilysin) | Cleaves C-terminal Phe-Arg (Pro-Phe bond) | Inactive fragments |
| Kininase I | Carboxypeptidase N (plasma), Carboxypeptidase M (tissue) | Removes C-terminal Arg → des-Arg⁹-bradykinin | Active B1R agonist |
| Aminopeptidase P | - | Cleaves N-terminal Arg¹-Pro² bond | Fragment susceptible to further cleavage |
| Dipeptidyl-peptidase IV | DPP-IV | Acts after aminopeptidase P cleavage | Inactive fragments |
Key clinical point: ACE (kininase II) inactivates bradykinin AND converts Ang I to Ang II. ACE inhibitors therefore block bradykinin degradation and reduce Ang II simultaneously - both mechanisms lower blood pressure. The accumulation of bradykinin is what causes the dry cough (and angioedema) seen with ACE inhibitors.
7. The KKS Cascade Diagram
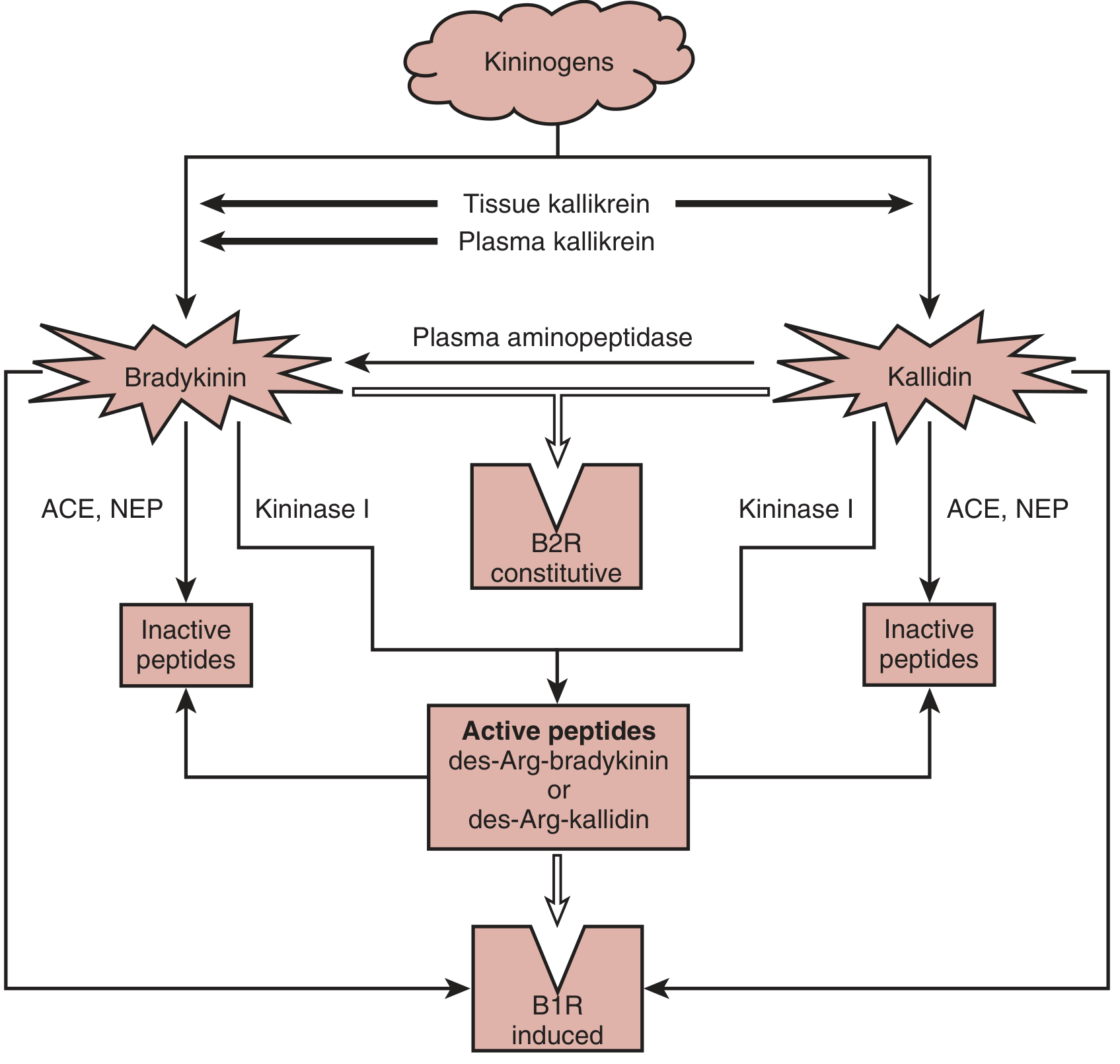
Fig. 11.8 from Brenner & Rector's The Kidney - Enzymatic cascade of the KKS. ACE = angiotensin-converting enzyme; B1R = bradykinin B1 receptor; B2R = bradykinin B2 receptor; NEP = neutral endopeptidase (neprilysin)
8. Physiological Effects of Kinins
Cardiovascular
- Vasodilation and hypotension: bradykinin infusion causes vasodilation via B2R on endothelial cells → NO + prostacyclin + hyperpolarizing EETs
- Animals deficient in tissue kallikrein show impaired flow-dependent vasodilation and endothelial dysfunction
- Cardioprotective: bradykinin contributes to ischemic preconditioning; stimulates tPA release from vascular endothelium (fibrinolysis)
- Many beneficial effects of ACE inhibitors are attributed to bradykinin accumulation (antiproliferative, anti-hypertrophic, improved insulin sensitivity)
- Goodman & Gilman's
Renal (Renal KKS)
- Kallikrein is produced in high amounts by connecting tubule epithelium - closely associated anatomically with afferent arterioles of the JGA
- Functions: regulates RBF, GFR, renin release, natriuresis, and diuresis
- Kinins influence tubuloglomerular feedback via prostaglandin production
- Modulates other vasoactive systems: renin-angiotensin, eicosanoids, catecholamines, NO, vasopressin, endothelin
- Urinary kallikrein is a marker of renal KKS activity; decreased in hypertension
- Brenner and Rector's The Kidney
Inflammation and Pain
- Kinins are potent mediators of inflammation: cause vasodilation, increase vascular permeability, edema
- Pain: bradykinin is one of the most potent endogenous pain-producing substances - activates sensory nociceptors (B1R and B2R)
- Stimulate prostaglandin synthesis in many tissues
Other Effects
- Smooth muscle contraction (airways, gut)
- Stimulates electrolyte and water transport across epithelia
- Cellular growth regulation
9. KKS and Other Systems - Intersections
| System | Interaction |
|---|---|
| Renin-Angiotensin System | ACE degrades both bradykinin and generates Ang II - opposing effects on BP; renal KKS is anatomically associated with JGA/renin cells |
| Coagulation (Contact System) | Plasma prekallikrein + HMW kininogen bind Factor XII on surfaces; activating the intrinsic coagulation pathway |
| Complement | Plasma kallikrein cleaves complement components |
| Prostaglandins | Kinins stimulate PLA₂ → arachidonic acid → PGs and EETs; many kinin effects are prostaglandin-mediated |
| Nitric Oxide | Kinins → eNOS activation (B2R) and iNOS (B1R in inflammation) |
10. Clinical Relevance and Pharmacology
Hereditary Angioedema (HAE)
- Caused by C1-inhibitor (C1-INH) deficiency; C1-INH normally inhibits plasma kallikrein and Factor XIIa
- Without C1-INH, uncontrolled kallikrein activation → massive bradykinin generation → angioedema
- Treatments targeting the KKS:
- Icatibant (Firazyr): selective competitive B2R antagonist; decapeptide; self-administered subcutaneously - Katzung
- C1-INH concentrates (Berinert, Cinryze): replace the missing inhibitor
- Ecallantide: recombinant plasma kallikrein inhibitor; more potent and selective than C1-INH
- Lanadelumab: monoclonal antibody inhibiting plasma kallikrein; HAE prophylaxis
- Donidalorsen: modified antisense oligonucleotide reducing plasma prekallikrein production
ACE Inhibitor Cough and Angioedema
- Bradykinin accumulation due to blocked kininase II → dry cough in ~10-15% of patients
- Rare but serious: bradykinin-mediated angioedema (does NOT respond to antihistamines/epinephrine - requires icatibant or C1-INH)
Inflammation and Pain Targets
- B2R antagonists (e.g., HOE-140): research tools; investigated for inflammatory pain
- Oral nonpeptide B2R antagonists (FR173657, NPC 18884): inhibit bradykinin-induced bronchoconstriction and nociception in animal models
- B1R antagonists (SSR240612): anti-inflammatory and analgesic in animal models
- Aprotinin: broad kallikrein inhibitor (used historically in cardiac surgery to reduce bleeding)
- Aspirin/NSAIDs: block prostaglandin-mediated kinin effects nonspecifically
Hypertension
- Urinary kallikrein is decreased in hypertension; kininogen-deficient rat strains show increased salt/angiotensin pressor sensitivity
- KKS appears minor in normal BP regulation but more important in hypertensive states
Cardiovascular Disease
- Selective B2R agonists under investigation for hypertension, myocardial hypertrophy
Summary
Prekallikrein (plasma, liver-derived)
↓ Factor XIIa (contact activation)
Plasma Kallikrein
↓ cleaves HMW kininogen
Bradykinin (B2R → vasodilation, ↑permeability, pain)
↓ Kininase I (Carboxypeptidase N/M)
des-Arg⁹-bradykinin (B1R → inflammation)
↓ Kininase II (ACE / Neprilysin)
Inactive peptides
Tissue Kallikrein (KLK1, local synthesis)
↓ cleaves LMW kininogen
Kallidin (B2R → same as bradykinin + local effects)
↓ Aminopeptidase
Bradykinin (interconversion)
Sources: Brenner and Rector's The Kidney | Goodman & Gilman's Pharmacological Basis of Therapeutics | Katzung's Basic and Clinical Pharmacology 16e
Layon hypothesis
Finding Sources
Searching the Web
Lyon hypothesis X chromosome inactivation Mary Lyon 1961
Reading File
Reading File
Reading File
Reading File
Loading Image
I have comprehensive material. Let me compile the full, detailed answer.
The Lyon Hypothesis (X-Chromosome Inactivation / Lyonization)
Note: You likely meant the Lyon hypothesis (not "Layon") - named after English geneticist Mary Frances Lyon (1925-2014), who proposed it in her landmark 1961 paper in Nature: "Gene Action in the X-chromosome of the Mouse."
Historical Background
In 1961, Mary Lyon observed a mosaic pattern of coat coloration in female mice that were heterozygous for X-linked genes affecting fur color. She noticed that one X chromosome in female mouse cells appeared more condensed (heteropycnotic) than the other. Building on earlier cytological observations (Susumu Ohno's work on sex chromatin), she proposed that this condensed chromosome was transcriptionally inactivated.
The process of X-chromosome inactivation (XCI) is now widely called lyonization in her honor. - Emery's Elements of Medical Genetics and Genomics
The Core Postulates of the Lyon Hypothesis
The hypothesis consists of four key principles:
| Postulate | Description |
|---|---|
| 1. Inactivation | One of the two X chromosomes in each female somatic cell is inactivated |
| 2. Randomness | The choice of which X (maternal Xm or paternal Xp) is inactivated is random in each cell |
| 3. Permanence | Once inactivated, the same X chromosome remains inactive in all daughter cells (clonal maintenance) |
| 4. Dosage compensation | This mechanism equalizes X-linked gene expression between XX females and XY males |
The Process Illustrated
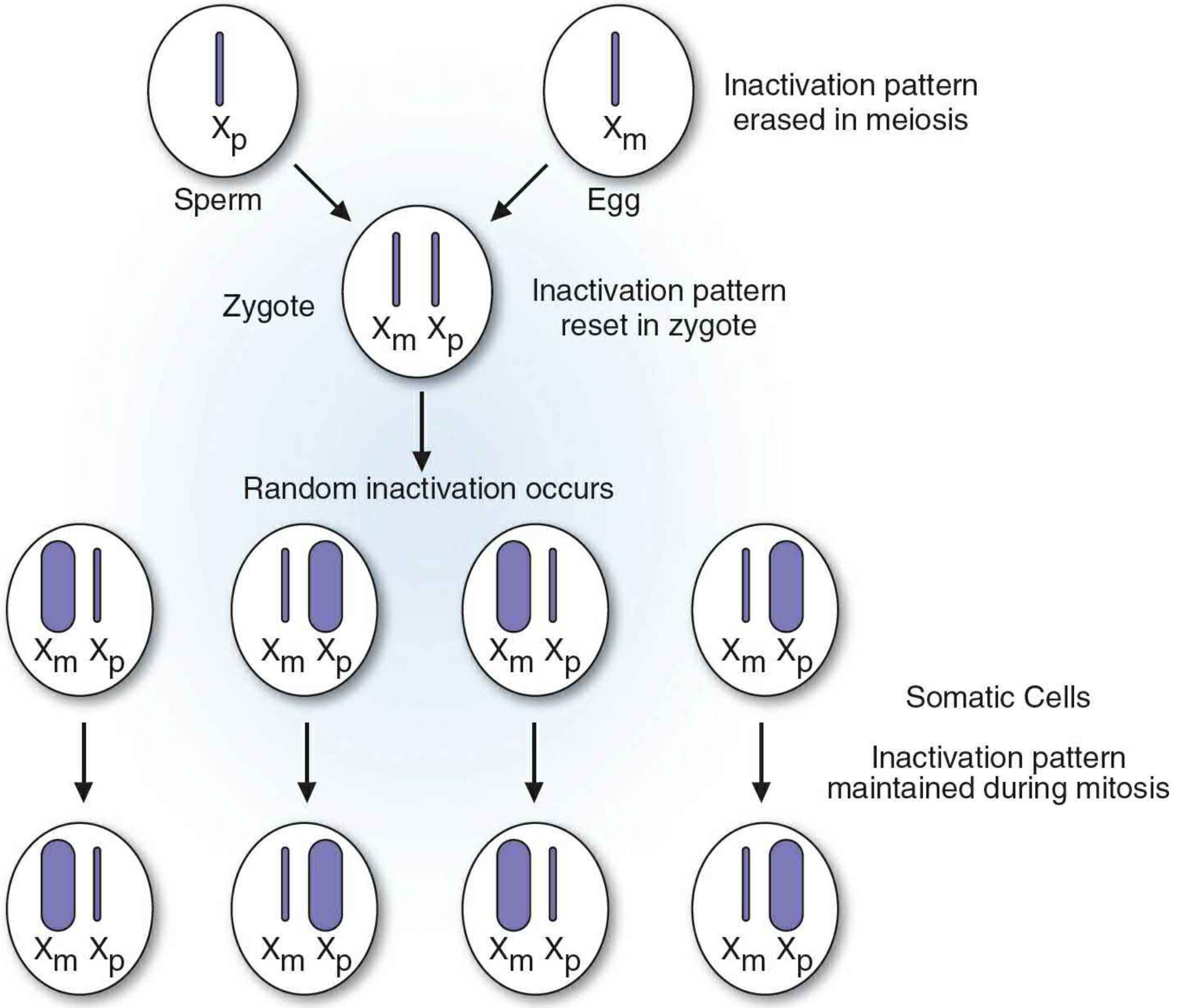
Figure 18.7 - The Lyon hypothesis. Inactivation is erased in the germ line, reset in the zygote, occurs randomly at the ~16-cell stage, and is then maintained clonally in all somatic progeny. - Basic Medical Biochemistry, 6e
Why Is Dosage Compensation Needed?
The X chromosome is approximately five times larger than the Y chromosome and contains ~900 genes. Without compensation, females (XX) would express the majority of X-linked genes at twice the level of males (XY). X inactivation ensures that both sexes have a single active X in each diploid somatic cell, regardless of how many X chromosomes are present. - Basic Medical Biochemistry, 6e; Thompson & Thompson
Timing of X Inactivation
- XCI begins at the earliest stages of embryogenesis - XIST RNA begins accumulating on one X chromosome around the 8-cell stage in human preimplantation embryos
- By approximately the 16-cell stage, inactivation is established and random
- Once set, the inactive X identity is mitotically stable and transmitted to all progeny cells
- In the germ line (oocytes), the inactive X is reactivated - so meiosis occurs with two active X chromosomes, ensuring that each egg carries one active X - Emery's; Basic Medical Biochemistry
Species difference: In humans and most placental mammals, inactivation is random (maternal or paternal X inactivated with equal probability). In marsupials, the paternally derived X chromosome is consistently inactivated (imprinted XCI). - Emery's
Molecular Mechanism of X Inactivation
The molecular machinery of XCI centers on the X Inactivation Center (XIC) at locus Xq13.3, which contains the critical gene XIST (X-Inactive Specific Transcript).
Steps:
- Counting: The cell "counts" the number of X chromosomes and inactivates all but one
- Choice: A stochastic process determines which X chromosome will be inactivated
- Initiation: The XIST gene on the future inactive X (Xi) begins to be expressed
- Spreading: XIST produces a long non-coding RNA (lncRNA) that "coats" the Xi in cis, spreading a methylation/silencing signal along the entire chromosome in both directions from the XIC
- Maintenance: Multiple epigenetic mechanisms lock in the silenced state:
Epigenetic Features Distinguishing Active X (Xa) vs. Inactive X (Xi): - Thompson & Thompson
| Feature | Active X (Xa) | Inactive X (Xi) |
|---|---|---|
| Gene expression | Yes; similar to male X | Most genes silenced; ~15% expressed to some degree |
| Chromatin state | Euchromatin | Facultative heterochromatin; Barr body |
| XIST RNA | XIST gene silenced | XIST RNA expressed from Xi only; coats the Barr body |
| DNA replication | Synchronous with autosomes | Late-replicating in S phase |
| Histone variant | Normal | Enriched for macroH2A |
| Histone modifications | Euchromatin marks | Enriched for heterochromatin marks (H3K27me3); deficient in euchromatin marks |
| DNA methylation | Hypomethylated promoters | Hypermethylated promoters at silenced genes |
The Barr Body
The inactive X chromosome condenses into a densely staining mass of sex chromatin visible at the nuclear periphery in interphase cells - known as the Barr body (named after Murray Barr, who first described it).
Rule: The number of Barr bodies = (total number of X chromosomes) - 1
| Karyotype | Sex | Barr bodies |
|---|---|---|
| 46,XY | Male | 0 |
| 46,XX | Female | 1 |
| 47,XXY (Klinefelter) | Male | 1 |
| 47,XXX | Female | 2 |
| 48,XXXY | Male | 2 |
| 48,XXXX | Female | 3 |
In extra X chromosome conditions (whether male or female), any X chromosome in excess of one is inactivated. - Emery's
Genes That Escape X Inactivation
Not all ~900 X-linked genes are silenced. Approximately 15% of X-linked genes show biallelic expression from both Xa and Xi to some degree. - Thompson & Thompson
- Pseudoautosomal regions (PAR1 and PAR2): At the tips of the X chromosome short and long arms; these regions are essentially identical on X and Y, undergo obligate recombination during male meiosis, and are not inactivated
- Distal Xp: Up to 50% of genes on distal Xp escape inactivation
- Xq: Only a few percent of genes escape inactivation
Clinical significance: Genes that escape inactivation explain why Turner syndrome (45,X), Klinefelter syndrome (47,XXY), and 47,XXX have clinical phenotypes - if all X genes were inactivated, extra or missing X chromosomes would be phenotypically silent. - Emery's
Females as Genetic Mosaics
Because XCI is random, all normal females are genetic mosaics - a mixture of two cell populations:
- Cells where the maternal X is active (Xm active, Xp inactive)
- Cells where the paternal X is active (Xp active, Xm inactive)
In healthy females, roughly 50% of cells express each X on average. However, the ratio can vary by chance or by selection.
Skewed X-Inactivation
Sometimes one X chromosome is preferentially inactivated, producing a non-random (skewed) pattern:
- If one X carries a lethal or severely deleterious mutation, cells with that X active will be eliminated by selection - the surviving cells preferentially have the normal X active
- Structural abnormalities: An abnormal X chromosome (translocations, deletions) is usually preferentially inactivated - only cells where the normal X is active survive
- Extreme skewing (>90:10) can allow a female carrier of an X-linked recessive disorder to become symptomatic (manifesting carrier)
- Emery's; Thompson & Thompson
Clinical Implications
1. X-Linked Recessive Disorders in Carrier Females
- Carrier females are usually unaffected because cells with the mutant X active are compensated by cells with the normal X active
- Extreme skewed inactivation toward the mutant X → manifesting carriers (e.g., mild hemophilia A, mild Duchenne muscular dystrophy, red-green color blindness in some female carriers)
2. X-Linked Dominant Disorders
- Males with only one X are often more severely affected or lethal (e.g., incontinentia pigmenti, Rett syndrome - males often do not survive)
- Females are more mildly affected due to mosaicism
3. Skewed XCI in Disease Detection
- Methylation-sensitive probes detecting XIST methylation patterns allow carrier detection for some X-linked diseases (e.g., Wiskott-Aldrich syndrome, X-linked severe combined immunodeficiency) - Emery's
4. ACE Inhibitors and XCI
- The XIST-based DNA methylation pattern has been exploited in clonality assays - useful for determining whether tumors or hematopoietic disorders (e.g., polycythemia vera, CML) are clonal expansions
5. Turner Syndrome (45,X)
- Only one X present → no Barr body, no XCI needed
- Clinical features (short stature, gonadal dysgenesis, lymphedema) arise from haploinsufficiency of genes that normally escape XCI and require two active copies (especially genes on Xp)
Key Summary
Zygote (XmXp) - both X chromosomes active
↓ ~8-16 cell stage
Random choice: Xm or Xp inactivated in each cell
↓
XIST lncRNA expressed from Xi → coats Xi → spreads silencing
↓
Epigenetic marks lock in Xi: hypermethylation, macroH2A, late replication, heterochromatin
↓
Clonally maintained in all daughter cells → mosaic female
↓
~15% of X genes escape; PAR genes always escape
The result: Each diploid somatic cell in both sexes has exactly one active X chromosome - achieving dosage compensation while creating female somatic mosaicism.
Sources: Thompson & Thompson Genetics and Genomics in Medicine, 9e | Emery's Elements of Medical Genetics and Genomics | Basic Medical Biochemistry: A Clinical Approach, 6e
This is a shared conversation. Sign in to Orris to start your own chat.