Sturg weber syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Sturge-Weber syndrome"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Here is the full, organized response:
Sturge-Weber Syndrome (Encephalotrigeminal Angiomatosis)
Overview
Sturge-Weber syndrome (SWS) is a sporadic neurocutaneous disorder (phacomatosis) characterized by the triad of:
- Facial port-wine nevus (capillary malformation)
- Ipsilateral leptomeningeal angioma
- Ocular abnormalities (particularly glaucoma)
It occurs in approximately 1 in 20,000 births and affects all races equally. It is not inherited - it is sporadic.
Etiology & Pathogenesis
SWS results from a somatic mosaic activating mutation in GNAQ (chromosome 9q21), with some cases also implicating GNA11. This mutation disrupts the q class of G-protein alpha subunits, reducing GTPase activity and increasing cell signaling. Because the mutation is somatic (not germline), it is not detectable on blood-based genetic testing - diagnosis rests on clinical findings.
Pathophysiologically, SWS results from persistence of the primitive embryonal vascular plexus that normally develops around the cephalic neural tube and facial skin during the 6th fetal week and regresses by the 9th week. When this regression fails, the persistent vascular plexus forms the characteristic angiomas.
Clinical Features
Cutaneous (Port-Wine Stain / Nevus Flammeus)

Two patients showing the classic port-wine nevus distribution. A: Nodular, thickened texture in an adult. B: Periorbital and episcleral involvement in a child - Bradley and Daroff's Neurology in Clinical Practice
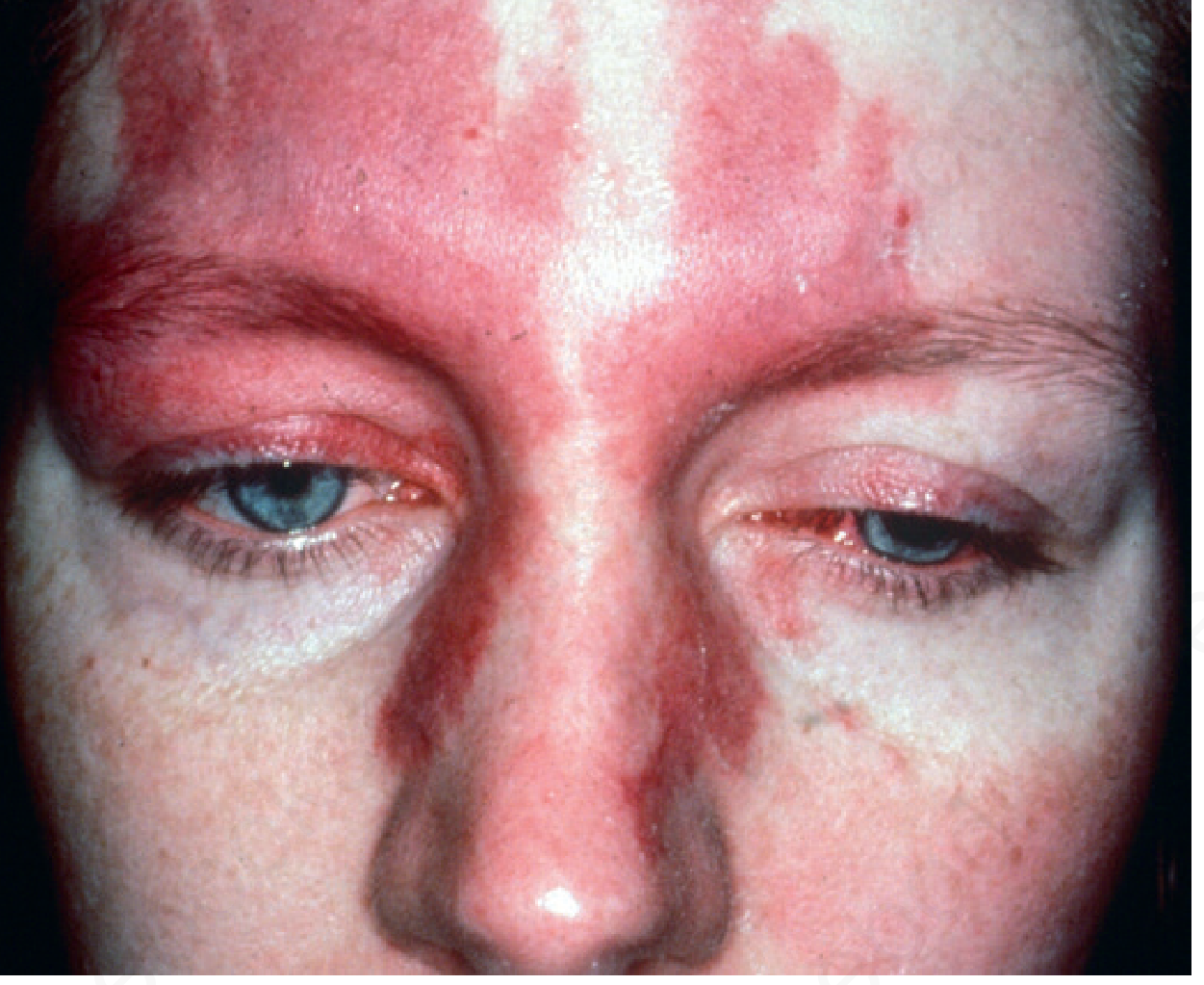
Bilateral capillary malformation (port-wine stain) with Sturge-Weber syndrome - Andrews' Diseases of the Skin
- The nevus involves the forehead and upper eyelid (V1/S1 distribution), may extend to lower face, trunk, and limbs
- Present at birth; may thicken and develop nodular texture over time
- Reactive hypertrophy of adjacent bone and connective tissue can occur
- Only 10-20% of children with a forehead port-wine nevus have a leptomeningeal angioma
- The more of the upper face involved, the higher the risk of intracranial involvement
- Bilateral V1 or V1/V2/V3 involvement significantly increases the risk of SWS
- Rarely, patients have the neurological/radiographic features of SWS without visible skin lesions
Neurological Features
Epilepsy, intellectual disability, and focal neurological deficits are the principal neurological abnormalities:
| Feature | Details |
|---|---|
| Epilepsy | Develops in 72-80% of patients with unilateral lesions, 93% with bilateral involvement |
| Seizure onset | 75% begin in the first year; 86% by age 2; 95% before age 5 |
| Seizure types | Focal motor, generalized tonic-clonic; also infantile spasms, myoclonic, atonic |
| Intellectual disability | Develops in ~50% overall; only 8% with bilateral brain involvement are intellectually normal |
| Hemiparesis | Often acute at onset of seizures; may be permanent (stroke-like) |
| Hemianopia | Visual field deficits common due to frequent occipital lobe involvement |
Early seizure onset (before age 2) is the strongest predictor of future intellectual disability and refractory epilepsy. Children with no seizures generally maintain normal intelligence. The condition eventually stabilizes, leaving residual deficits without further deterioration.
Ocular Features
- Glaucoma is the main ophthalmological complication, with two age peaks: infancy and late childhood
- Buphthalmos (enlarged globe) and amblyopia in some neonates
- Choroidal angioma (diffuse, ipsilateral)
- Episcleral hemangioma - causes blood in Schlemm's canal on gonioscopy
- Iris heterochromia (ipsilateral to nevus)
- Glaucoma can occur even without neurological involvement
- Periodic intraocular pressure measurement is mandatory whenever the nevus is near the eye
Radiology / Imaging
CT Scan
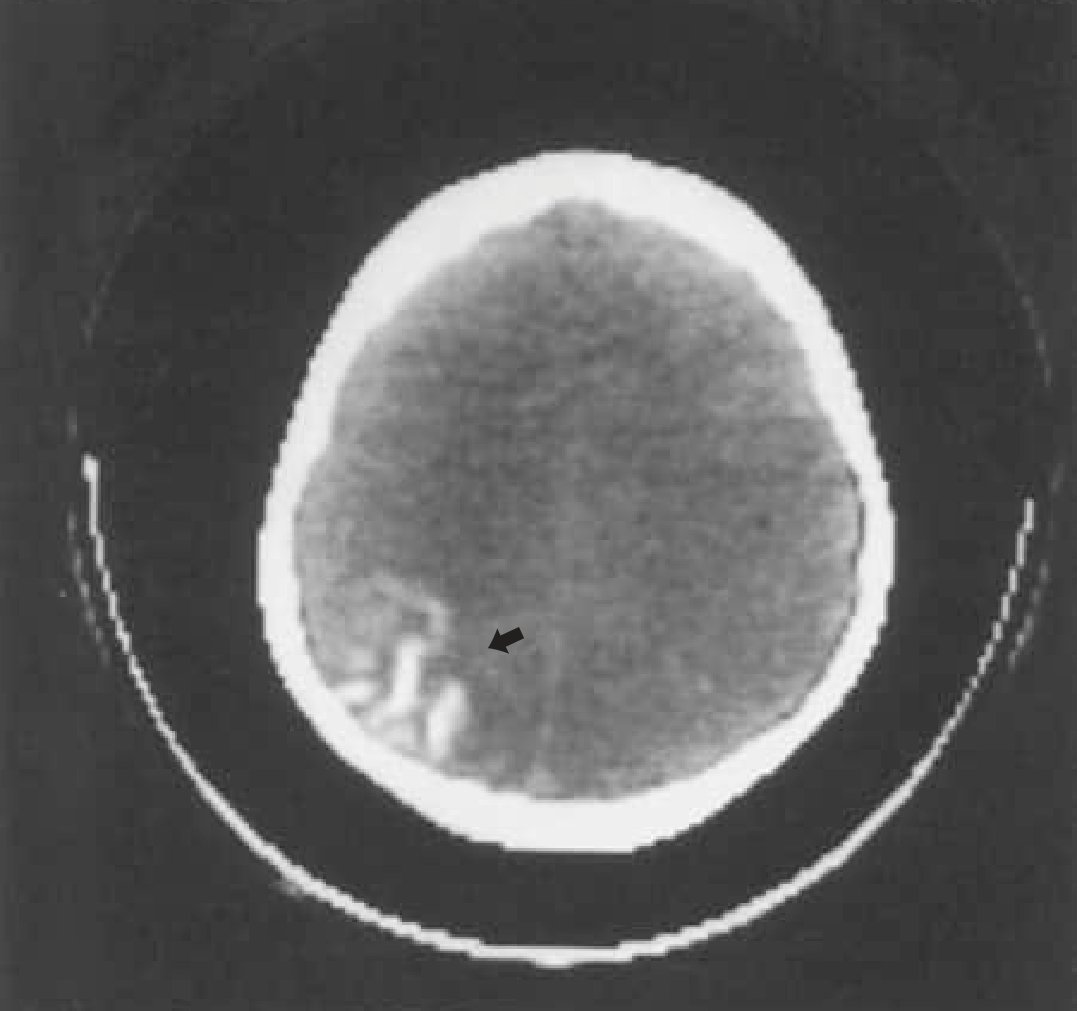
CT head showing the classic occipital gyriform "tram-track" calcification pattern (arrow) - Bradley and Daroff's Neurology
- Classic finding: "tram-track" or "tramline" calcifications following cortical gyri (calcium deposition in outer cortical layers)
- CT detects calcification earlier than plain skull radiographs
- Usually develops by age 2 years; almost never present in neonates
- Calcifications result from abnormal venous drainage causing chronic cortical ischemia
MRI
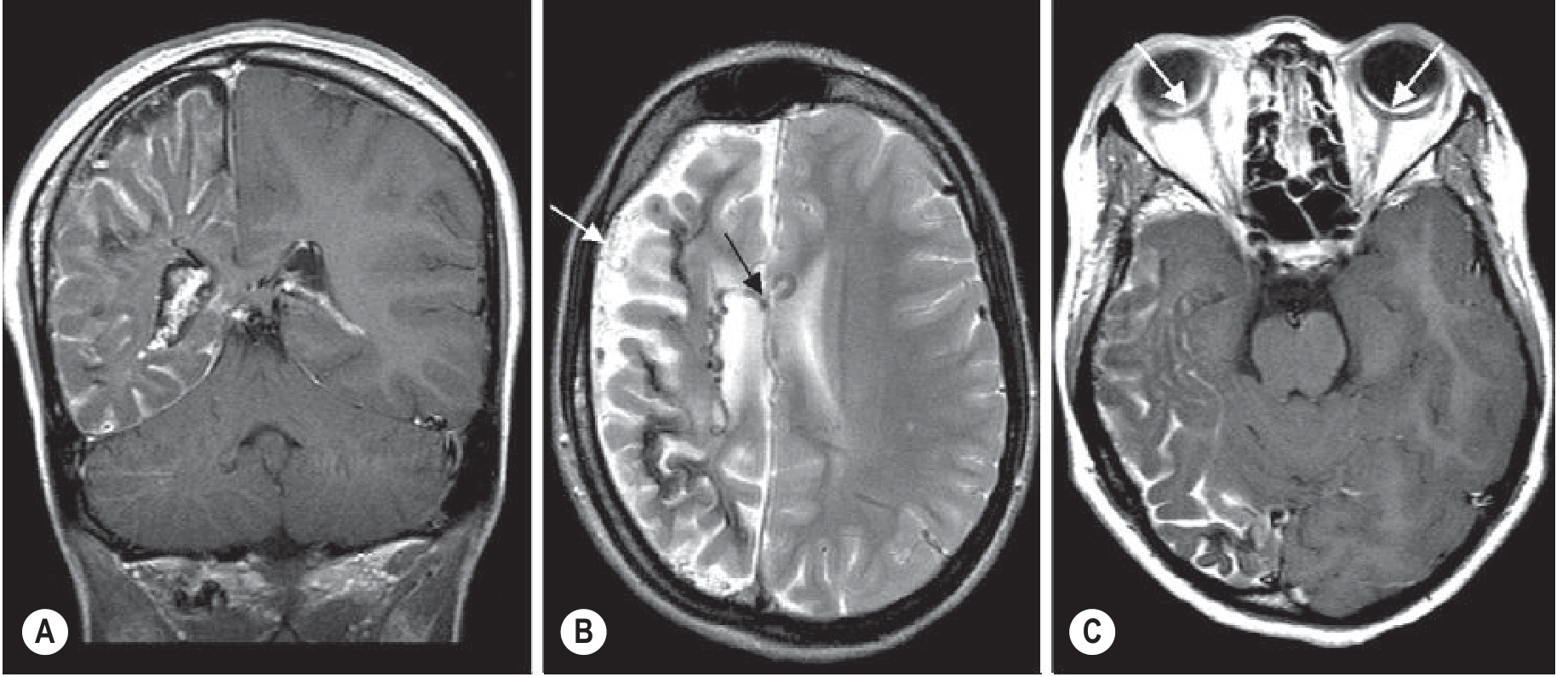
Sturge-Weber syndrome MRI: (A) Coronal T1 post-contrast - enhancing pial angioma over right hemisphere with cortical atrophy and enlarged choroid plexus. (B) Axial T2 - prominent superficial and ependymal veins (arrows). (C) Axial post-contrast T1 - bilateral choroidal angiomas (arrows) - Grainger & Allison's Diagnostic Radiology
- Gadolinium-enhanced MRI is the most sensitive investigation, especially in infancy before calcifications develop
- Findings include: diffuse pial enhancement (leptomeningeal angioma), enlarged ipsilateral choroid plexus, dilated transparenchymal veins, and progressive hemispheric atrophy
- T1 without contrast may appear completely normal early on
- In "burnt-out" cases, the angioma may no longer enhance - only a shrunken, calcified hemisphere remains
- MRI is preferred over CT in infancy; both are complementary
- PET shows reduced glucose metabolism adjacent to the lesion (may show increased metabolism with recent seizures)
- SPECT shows reduced perfusion, often revealing involvement beyond what CT shows
Classification (Roach Scale)
| Type | Features |
|---|---|
| Type I | Facial AND leptomeningeal angioma (classic SWS); may have glaucoma |
| Type II | Facial angioma only; no intracranial involvement |
| Type III | Isolated leptomeningeal angioma; no facial nevus |
Diagnosis
The diagnosis is clinical - the somatic mosaic GNAQ mutation is not detectable in peripheral blood. Neuroimaging is used to confirm intracranial involvement. MRI with gadolinium contrast is the preferred first investigation in an infant with a relevant port-wine stain.
Management
Seizures
- Antiepileptic drugs (AEDs) are first-line; however, seizures are often refractory
- Surgical options include focal cortical resection or hemispherectomy for refractory epilepsy - can improve seizure control and may promote better cognitive development
- Patient selection criteria: refractory seizures + clinical dysfunction in the resection area + failure of adequate AED trial
- Corpus callosotomy is used for refractory tonic or atonic seizures with extensive bilateral disease
Aspirin
- Low-dose aspirin is offered to reduce stroke-like episodes, seizures, and cognitive impairment
- Early institution may help prevent seizures and developmental delay (particularly in those confirmed to have SWS on imaging)
- Generally well-tolerated; efficacy not definitively established by RCTs
Glaucoma
- Requires periodic IOP monitoring from birth
- Medical and/or surgical treatment for elevated IOP
Port-Wine Stain
- Pulsed dye laser is the treatment of choice for lightening the nevus (ideally started early in infancy when lesions are more responsive)
Key Points to Remember
- SWS is a phacomatosis (neurocutaneous syndrome), alongside NF1, NF2, tuberous sclerosis, and Von Hippel-Lindau
- Caused by a somatic GNAQ mutation - sporadic, not inherited
- The classic triad: port-wine stain (V1) + leptomeningeal angioma + glaucoma
- Only 10-20% of upper facial port-wine stains have associated brain involvement
- Leptomeningeal angioma is usually ipsilateral but bilateral brain lesions occur in at least 15%
- MRI + gadolinium is the investigation of choice in infancy
- The classic CT appearance is "tram-track" gyriform calcification in parieto-occipital cortex
- Seizure onset before age 2 is the strongest predictor of intellectual disability
- Bilateral brain involvement is associated with much worse cognitive outcomes (only 8% intellectually normal)
Sources: Bradley and Daroff's Neurology in Clinical Practice | Goldman-Cecil Medicine | Grainger & Allison's Diagnostic Radiology | Andrews' Diseases of the Skin | Kanski's Clinical Ophthalmology
Recent evidence note: A 2024 systematic review (PMID 39043228) examined headache as a feature of SWS, noting it is an underrecognized manifestation. A 2025 systematic review (PMID 40447045) highlighted that diffuse leptomeningeal melanomatosis can mimic Type III SWS and should be considered in the differential.
This is a shared conversation. Sign in to Orris to start your own chat.