Congenital facial anomalies
Reading File
Finding Sources
Finding Sources
Searching PubMed
congenital facial anomalies classification management
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Now I have comprehensive material. Let me compile the full response.
Congenital Facial Anomalies
Congenital facial anomalies are structural defects of the face present at birth, arising from disturbances in the normal embryologic development of the facial prominences and pharyngeal arches. The face forms primarily between weeks 4-8 of gestation from five prominences: the unpaired frontonasal prominence and the paired maxillary and mandibular prominences derived from the first pharyngeal arch.
1. Embryologic Basis
At the end of week 4, neural crest-derived facial prominences emerge from the first pair of pharyngeal arches. The frontonasal prominence forms the forehead, nose, and central upper lip. The maxillary prominences form the lateral upper lip, cheeks, and secondary palate. The mandibular prominences fuse to form the lower jaw. Failure of fusion, deficient mesenchyme, or abnormal apoptosis at critical junctions underlies most facial clefts and structural anomalies.
Key molecular signals involved include FGF receptors (FGFR1, FGFR2), MSX2, TWIST, and ALX4 genes - mutations in these are linked to multiple syndromes.
2. Cleft Lip and Cleft Palate
The most common congenital facial anomaly, affecting approximately 1 in 1000 births in the United States (~7000 neonates/year). Males are affected in 60-80% of cases.
Anatomic Landmark
The incisive foramen is the critical landmark dividing:
- Primary palate - premaxilla, lip, nasal tip, and columella (anterior to incisive foramen)
- Secondary palate - from incisive foramen to uvula
Classification
Cleft Lip:
| Type | Description |
|---|---|
| Unilateral incomplete | Partial vertical height; may have Simonart band (skin bridge) or forme fruste |
| Unilateral complete | Full thickness; extends into nasal floor + alveolus |
| Bilateral | Both sides; median palatal process protrudes freely anteriorly |
Cleft Palate:
| Type | Description |
|---|---|
| Cleft uvula | Mildest form |
| Submucous cleft palate (SMCP) | Muscular diastasis with intact mucosa; bifid uvula, zona pellucida, posterior hard palate notch |
| Unilateral secondary palate cleft | Palatal process fused to nasal septum on one side only |
| Bilateral complete | No fusion between maxilla and nasal septum |
| Complete palatal cleft | Involves both primary and secondary palate; usually with cleft lip |
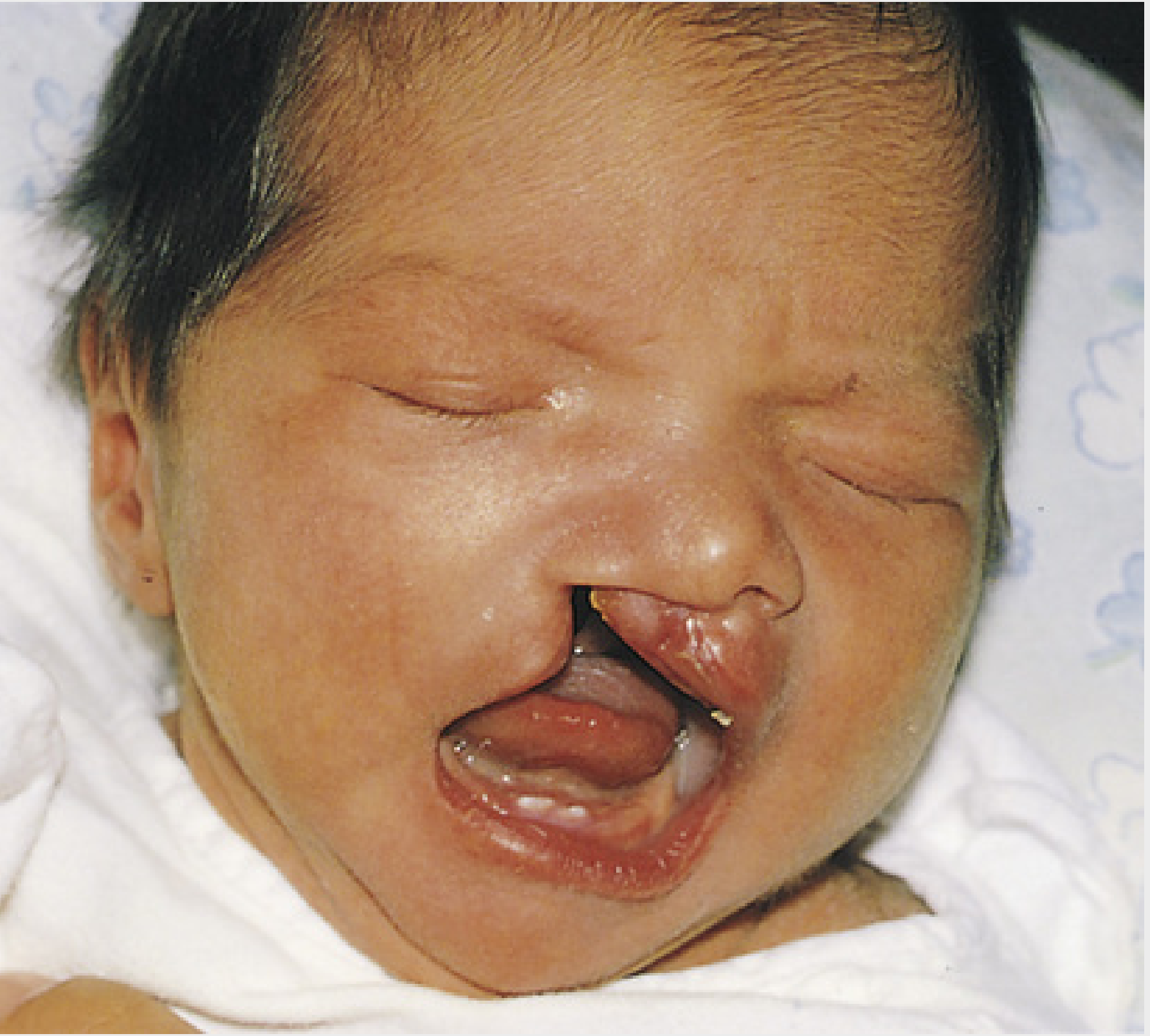
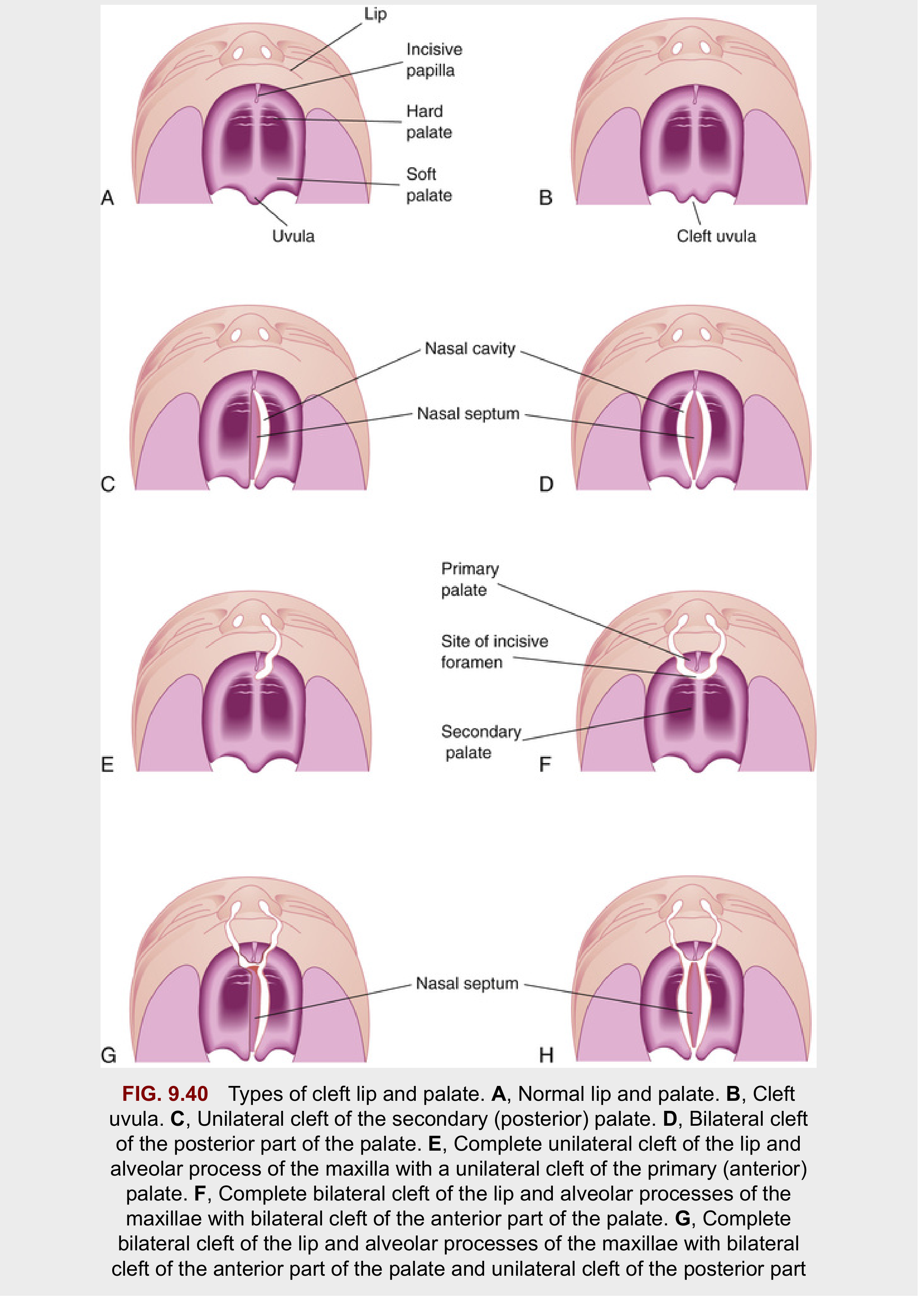
Etiology
- Unilateral cleft lip - failure of the maxillary prominence to unite with the merged medial nasal prominences on one side
- Bilateral cleft lip - same failure on both sides; the epithelium in the labial groove stretches and breaks down
- Cleft palate - failure of the lateral palatine processes (palatal shelves) to elevate, fuse, or undergo proper midline epithelial dissolution
- Risk factors: maternal valproic acid, folate deficiency, alcohol, smoking, corticosteroid use; genetic factors (chromosomal syndromes, single-gene disorders)
Consequences and Management
- Feeding difficulties (poor latch), recurrent otitis media, speech and hearing defects, dental malocclusion
- Lip repair (cheiloplasty): classically at 3 months ("rule of 10s" - 10 weeks, 10 lbs, 10 g Hb)
- Palate repair: 9-18 months to allow speech development before age-critical period
- Multidisciplinary team: plastic surgery, orthodontics, speech therapy, audiology, ENT, psychology
3. Craniosynostosis
Premature fusion of one or more cranial sutures, incidence 1:2500 births, more common in males. Mutations in MSX2, ALX4, FGFR1, FGFR2, and TWIST are implicated. Associated with maternal valproic acid use, smoking, and thyroid disease.
Suture closure prevents growth perpendicular to the suture, forcing compensatory growth parallel to it.
| Suture Fused | Skull Shape | Name |
|---|---|---|
| Sagittal (50% of cases) | Long, narrow, wedge-shaped | Scaphocephaly (dolichocephaly) |
| Bilateral coronal (30%) | High, tower-like | Brachycephaly |
| Unilateral coronal | Twisted, asymmetric | Plagiocephaly |
| Frontal (metopic) | Deformity of frontal/orbital bones | Trigonocephaly |
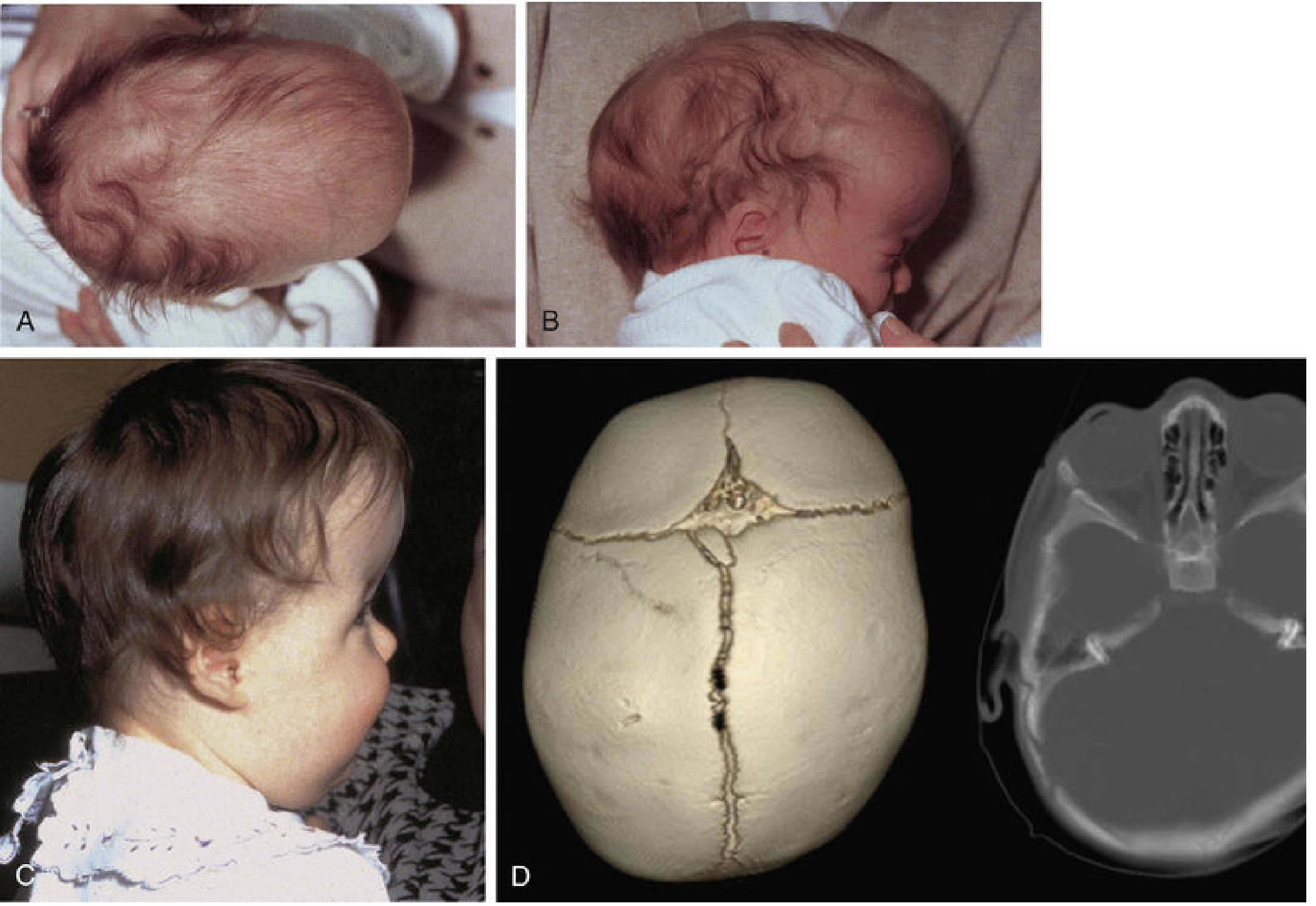
Note: Positional (deformational) plagiocephaly - the most common cranial deformity - results from repeated head positioning, not suture fusion. It does not require surgery; repositioning suffices.
Syndromic craniosynostoses involving the face include:
- Apert syndrome - FGFR2 mutation; acrocephaly, midface hypoplasia, syndactyly
- Crouzon syndrome - FGFR2 mutation; craniosynostosis, exorbitism, maxillary hypoplasia
- Pfeiffer syndrome - FGFR1/FGFR2 mutation; broad thumbs, proptosis
4. Treacher Collins Syndrome (Mandibulofacial Dysostosis)
- Genetics: Autosomal dominant; TCOF1 gene mutation; ~50% are new mutations; incidence 1:50,000 live births
- Pathology: Bilateral defects in first and second branchial arch derivatives
- Features:
- Down-slanting palpebral fissures
- Lower eyelid colobomas and eyelash/follicle malformations
- Absent or hypoplastic zygomatic bones
- Mandibular hypoplasia (retrognathia, steep occlusal plane)
- Bilateral microtia/anotia; hypoplasia or atresia of external auditory meatus
- Conductive hearing loss (inner ear usually spared)
- Cleft palate in 35% of cases
- Intelligence is typically normal

Management: Immediate airway management in neonates (may require tracheostomy); hearing aids; multistage craniofacial reconstruction; orthodontics.
5. Hemifacial Microsomia (Oculoauriculovertebral Spectrum / Goldenhar Syndrome)
- Incidence: 1:5600 births; usually sporadic
- Hypothesized etiology: unilateral hemorrhagic event involving the stapedial artery during early development
- Features (unilateral or occasionally bilateral):
- Temporal, zygomatic, maxillary, and mandibular hypoplasia
- Hypoplastic facial musculature; facial paresis/paralysis
- External and middle ear deformities (aural atresia, preauricular skin tags)
- Ocular involvement: colobomas, epibulbar dermoids (choristomas), blepharophimosis, strabismus
- Cleft lip/palate, vertebral anomalies (fused vertebrae, spina bifida)
The condition spans a spectrum - at one extreme: mild unilateral lower lip paralysis; at the other extreme: complete facial paralysis with severe bony hypoplasia.
6. Holoprosencephaly and Midline Facial Defects
These result from failure of the forebrain (prosencephalon) to divide into two hemispheres, often accompanied by midline facial defects due to loss of midline tissue as early as days 19-21 of gestation.
| Severity | Brain | Face |
|---|---|---|
| Alobar (most severe) | Single undivided hemisphere | Cyclopia (single midline eye), proboscis, severe hypotelorism |
| Semilobar | Partial division | Hypotelorism, midline cleft lip |
| Lobar (least severe) | Near-complete division | Hypotelorism, single central incisor |
Genetic causes include SHH, ZIC2, SIX3, and PTCH1 mutations, as well as trisomy 13 (Patau syndrome).
7. Summary Table of Key Congenital Facial Anomalies
| Anomaly | Key Features | Genetics/Cause | Incidence |
|---|---|---|---|
| Cleft lip ± palate | Orofacial cleft, speech/feeding defects | Multifactorial; folate deficiency, teratogens | 1:1000 |
| Cleft palate alone | Isolated secondary palate; velopharyngeal insufficiency | Multifactorial | ~1:2000 |
| Craniosynostosis | Premature suture fusion, skull deformity | FGFR, MSX2, TWIST mutations | 1:2500 |
| Treacher Collins | Bilateral malar/mandibular hypoplasia, microtia | TCOF1 (AD) | 1:50,000 |
| Hemifacial microsomia | Unilateral face/ear hypoplasia, epibulbar dermoid | Sporadic (stapedial artery) | 1:5600 |
| Holoprosencephaly | Cyclopia/midline defects, forebrain non-division | SHH, trisomy 13 | Variable |
| Apert/Crouzon | Craniosynostosis + midface hypoplasia | FGFR2 (AD) | Rare |
8. Multidisciplinary Management Principles
All congenital facial anomalies benefit from a dedicated craniofacial team including:
- Craniofacial/plastic surgeon
- Oral and maxillofacial surgeon
- Orthodontist and pediatric dentist
- Audiologist and ENT surgeon
- Speech-language pathologist
- Ophthalmologist
- Clinical geneticist
- Psychologist/social worker
Surgical timing follows growth-stage principles to avoid disturbing active facial growth centers, with most major skeletal corrections deferred until skeletal maturity (mid-teens), while functional corrections (airway, feeding, hearing) are addressed early.
Sources: The Developing Human - Clinically Oriented Embryology; Cummings Otolaryngology Head and Neck Surgery; Scott-Brown's Otorhinolaryngology Head & Neck Surgery (Vol. 2); Adams and Victor's Principles of Neurology, 12th ed.
This is a shared conversation. Sign in to Orris to start your own chat.