Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Searching Images
ARDS acute respiratory distress syndrome pathophysiology diffuse alveolar damage mechanism diagram
Loading Image
Loading Image
I now have all the material needed for a thorough, well-illustrated answer. Here is the comprehensive explanation:
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition and Overview
ARDS is a syndrome of noncardiogenic pulmonary edema characterized by severe lung inflammation, diffuse alveolar damage (DAD), hypoxemia, and reduced lung compliance leading to respiratory failure. Unlike cardiogenic pulmonary edema (caused by elevated hydrostatic pressures), the edema in ARDS is exudative — the alveolar-capillary barrier breaks down and protein-rich fluid leaks into the airspaces.
"In ARDS, the edema fluid that fills the alveoli is exudative. In other words, the alveolar-capillary barrier exhibits increased permeability, allowing for the leakage of protein-rich fluid into the airspaces." — Murray & Nadel's Textbook of Respiratory Medicine
Triggers: Direct vs. Indirect Injury
| Direct (Pulmonary) | Indirect (Nonpulmonary) |
|---|---|
| Pneumonia (bacterial, viral, COVID-19) | Sepsis |
| Aspiration | Major trauma |
| Pulmonary contusion | Multiple blood transfusions |
| Toxic inhalation / near-drowning | Pancreatitis, cardiopulmonary bypass |
| Reperfusion injury (post-transplant) | Drug overdose |
Whether the trigger is pulmonary or nonpulmonary, the downstream lung pathology is indistinguishable.
Core Pathophysiological Mechanisms
1. Alveolar-Capillary Barrier Disruption
The alveolar-capillary unit has two cellular layers that normally keep the alveoli dry:
- Pulmonary microvascular endothelium (capillary side)
- Alveolar epithelium — type I pneumocytes (gas exchange, ~95% of surface area) and type II pneumocytes (surfactant production, progenitor cells)
In ARDS, both layers are injured. Type I pneumocyte destruction is particularly critical because these cells are largely non-regenerative. Type II pneumocyte injury impairs surfactant production, further collapsing alveoli.
The result: loss of the fluid exclusion barrier → protein-rich fluid, fibrin, and cellular debris flood the alveolar space.
2. Neutrophil-Mediated Injury (Central Mechanism)
Neutrophils are recruited in massive numbers from the pulmonary microvasculature into the alveolar space and are the primary effectors of DAD. They cause injury via:
- Reactive oxygen species (ROS) — oxidative damage to lipid membranes and proteins
- Proteases (elastase, matrix metalloproteinases) — degrade the extracellular matrix and tight junction proteins
- Neutrophil extracellular traps (NETs) — chromatin + enzymes that amplify inflammation and activate complement and coagulation
- Myeloperoxidase (MPO) — generates hypochlorous acid, further damaging epithelium
Neutrophil recruitment is driven by chemokines (particularly IL-8/CXCL8) and platelet-activating factor (PAF) released from activated alveolar macrophages and endothelial cells.
3. The Cytokine Storm
Alveolar macrophages (the first-line responders) detect danger signals via TLR/PRR pathways and release a cascade of pro-inflammatory mediators:
| Mediator | Role |
|---|---|
| TNF-α, IL-1β | Activate endothelium, increase adhesion molecules (ICAM-1, E-selectin), recruit neutrophils |
| IL-6 | Amplifies inflammation; elevated levels predict mortality |
| IL-8 (CXCL8) | Primary neutrophil chemoattractant into the lung |
| IL-17 | Released by Th17 cells; amplifies neutrophilic inflammation |
| Prostaglandins, leukotrienes | Increase vascular permeability |
| Complement fragments (C3a, C5a) | Further recruit and activate neutrophils; increase permeability |
| Angiopoietin-2 (Ang-2) | Destabilizes endothelial junctions; elevated Ang-2 predicts mortality |
The T-regulatory cell population is simultaneously suppressed, removing a key brake on the inflammatory response.
4. Endothelial and Epithelial Tight Junction Breakdown
Cytokines and proteases disrupt intercellular tight junctions (occludin, claudins, VE-cadherin on endothelial cells), causing a loss of paracellular barrier function. Fluid, plasma proteins, and leukocytes pour from capillaries into the interstitium and then into alveoli. This is fundamentally different from hydrostatic edema — the protein content of the edema fluid is nearly equal to plasma.
5. Surfactant Dysfunction
- Type II pneumocyte destruction reduces surfactant synthesis
- Plasma proteins (especially fibrinogen) that flood the alveoli inactivate existing surfactant
- Phospholipase A2 (released during pancreatitis or systemic inflammation) directly degrades surfactant phospholipids
Without surfactant, alveolar surface tension rises dramatically → alveolar collapse (atelectasis) and severely reduced lung compliance ("stiff lungs").
6. Coagulation Dysregulation
The injured alveolar microenvironment activates the coagulation cascade:
- Fibrin deposition in alveoli contributes to hyaline membrane formation
- Microvascular thrombosis reduces capillary perfusion
- Simultaneous impairment of fibrinolysis (through elevated PAI-1) prevents clot resolution
- This creates a procoagulant, antifibrinolytic environment within the lung
7. Pulmonary Hypertension
Multiple mechanisms contribute to elevated pulmonary artery pressure in ARDS:
- Hypoxic pulmonary vasoconstriction (the Euler-Liljestrand reflex, usually protective, becomes maladaptive when widespread)
- Intravascular fibrin/thrombus obstructing pulmonary arterioles
- External compression of vessels by positive-pressure ventilation
- Mediator-driven vasoconstriction (thromboxane A2, ET-1)
The Three Histopathological Phases
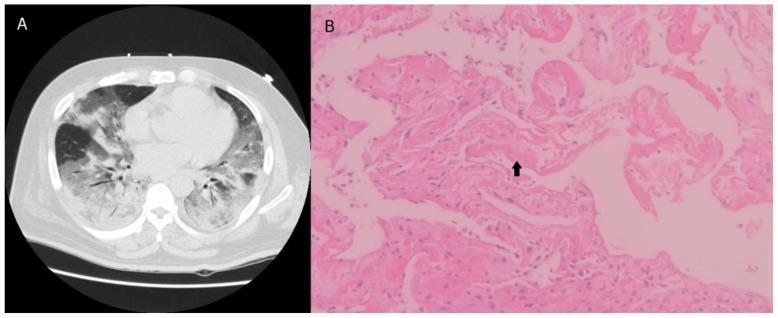
Phase 1: Exudative (Days 1–7)
- Protein-rich edema fluid, fibrin, and neutrophils flood the alveoli
- Hyaline membrane formation (cellular debris + fibrin lining the alveolar walls) — the histological hallmark
- Widespread type I pneumocyte necrosis
- Interstitial infiltration with neutrophils
- Clinical: severe hypoxemia, bilateral infiltrates on imaging
Phase 2: Proliferative (Days 7–21)
- Type II pneumocytes proliferate to cover denuded alveolar walls (attempting repair)
- Organization of hyaline membranes
- Early fibroblast infiltration → early fibrosis
- Decline in neutrophils
- Some patients improve; others progress
Phase 3: Fibrotic (>21 days)
- Progressive pulmonary fibrosis in a subset of patients
- Obliteration of alveolar spaces and pulmonary capillaries
- Collagen deposition in alveoli and interstitium
- Elevated N-terminal procollagen peptide III in BAL fluid can be detected as early as 24 hours, indicating early fibrogenesis
- Associated with prolonged mechanical ventilation and worse prognosis
Physiological Consequences
| Consequence | Mechanism |
|---|---|
| Severe hypoxemia (refractory) | Right-to-left shunt through fluid/collapsed alveoli; no response to supplemental O₂ alone |
| Reduced lung compliance | Alveolar flooding, atelectasis, surfactant loss → "stiff lungs" |
| Increased dead space | Microvascular occlusion → ventilated but unperfused alveoli → ↑ minute ventilation required |
| Pulmonary hypertension | Vasoconstriction, microvascular thrombosis, compression |
| Multiorgan dysfunction | Cytokine spillover into systemic circulation; ventilator-induced lung injury further amplifies mediator release (biotrauma) |
Ventilator-Induced Lung Injury (VILI) — A Secondary Mechanism
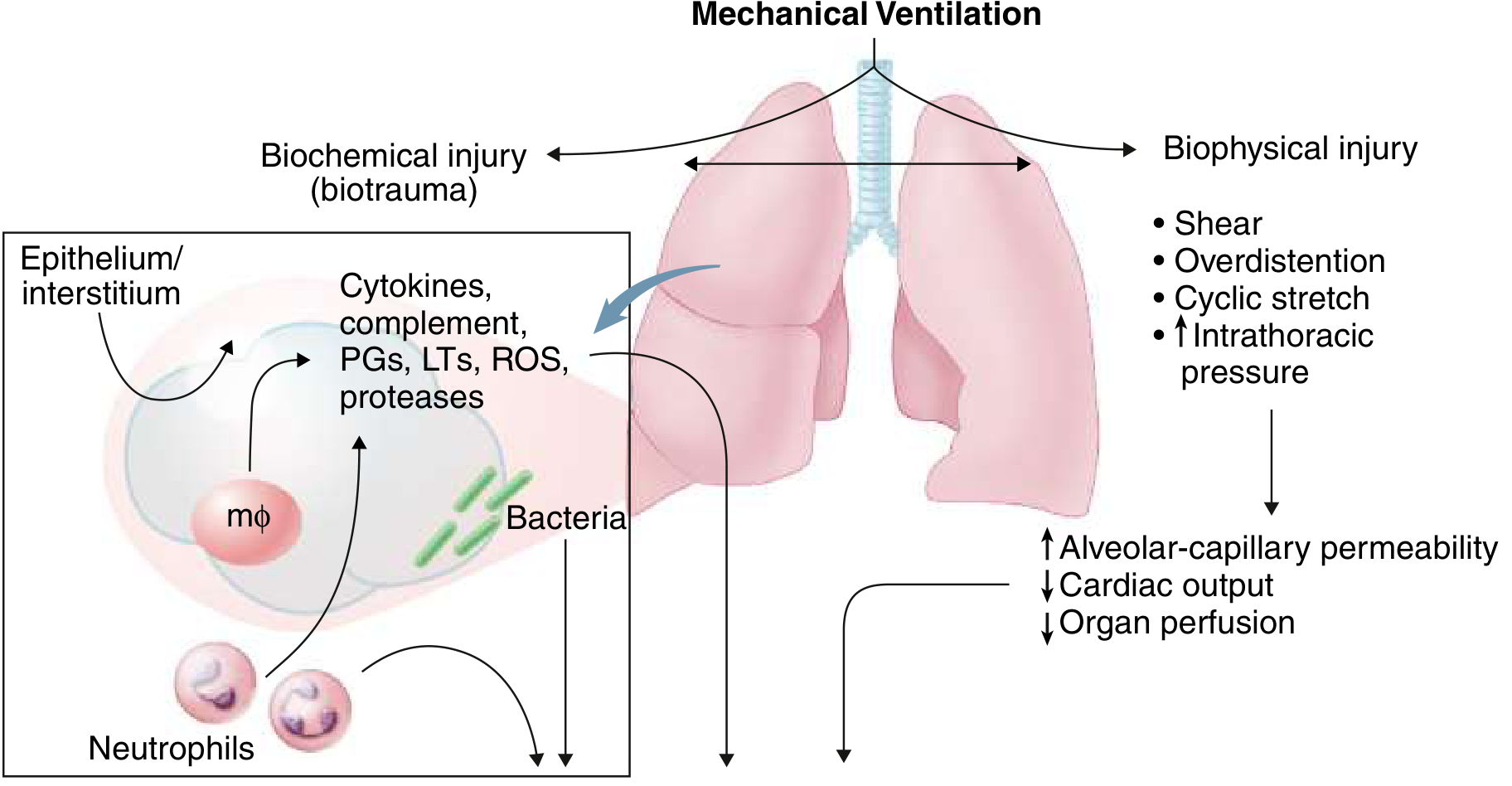
Mechanical ventilation itself worsens ARDS through:
- Volutrauma/barotrauma: Overdistension of remaining aerated alveoli
- Atelectrauma: Repetitive collapse-reopening cycles at end-expiration
- Biotrauma: Mechanical stretch activates lung macrophages and epithelial cells, triggering further cytokine release that spills systemically — contributing to multiorgan failure
This is the physiological rationale for lung-protective ventilation (low tidal volumes 6 mL/kg predicted body weight, plateau pressure <30 cmH₂O).
Summary Cascade
Triggering insult (sepsis, pneumonia, trauma, aspiration...)
↓
Alveolar macrophage activation → TLR/PRR signaling
↓
Cytokine storm (TNF-α, IL-1β, IL-6, IL-8, complement)
↓
Endothelial activation → neutrophil adhesion & transmigration
↓
Neutrophil-mediated injury (ROS, proteases, NETs, MPO)
↓
Alveolar-capillary barrier breakdown
↓
Protein-rich exudative edema floods alveoli
↓
Surfactant inactivation + alveolar collapse
↓
Hyaline membrane formation (DAD) + coagulation activation
↓
Hypoxemia, reduced compliance, pulmonary hypertension
↓
ARDS → potential fibrosis → multiorgan dysfunction
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter 134 (Pathogenesis and Pathology of ARDS)
- Goldman-Cecil Medicine, Chapter 91 (ARDS and Mechanical Ventilation)
- Goldman-Cecil Medicine, Chapter 90
This is a shared conversation. Sign in to Orris to start your own chat.