Define hsp from basics to advance clinical
Reading File
Finding Sources
Finding Sources
Searching PubMed
"IgA vasculitis" OR "Henoch-Schonlein purpura"
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Loading Image
I now have comprehensive data from 7 major textbooks. Let me compile the full, detailed answer.
IgA Vasculitis (HSP) - Basics to Advanced Clinical
1. Nomenclature and Definition
Henoch-Schonlein Purpura (HSP) is now formally termed IgA Vasculitis (IgAV) under the 2012 revised Chapel Hill Consensus Conference nomenclature. The eponym HSP refers to Eduard Heinrich Henoch and Johann Lukas Schonlein, who described the clinical tetrad in the 19th century. The name change reflects the defining pathophysiologic feature: IgA immune complex deposition in blood vessel walls.
IgA vasculitis is a small-vessel vasculitis characterized by palpable purpura (most commonly distributed over the buttocks and lower extremities), arthralgias, gastrointestinal signs and symptoms, and glomerulonephritis.
- Harrison's Principles of Internal Medicine 22E
2. Epidemiology
| Parameter | Details |
|---|---|
| Most common vasculitis in children | Yes - 10-20 per 100,000/year in children |
| Peak age (children) | 4-7 years (range 2-8 years) |
| Children vs adults | ~75% of cases in children |
| Average adult onset age | ~50 years |
| Sex ratio | Male:female = 1.5:1 |
| Seasonal pattern | Peak in spring; rare in summer |
| Recurrence | 5-30% of patients |
Adults may also be affected across a broad age range and tend to have a more prolonged disease course with recurrent bouts of purpura and a higher rate of severe renal complications.
- ROSEN's Emergency Medicine; Andrews' Diseases of the Skin
3. Pathogenesis - From Molecular to Clinical
This is where the "basics" connect to the mechanism:
Step 1: Aberrant IgA1 Glycosylation (The Core Defect)
A fraction of circulating IgA1 molecules has aberrant O-linked glycosylation in the hinge region. Normally, O-glycans terminate with galactose. In IgAV (and IgA nephropathy), the glycans are galactose-deficient (Gd-IgA1) - they end with N-acetylgalactosamine (GalNAc) or sialylated GalNAc instead.
Step 2: Anti-glycan Autoantibodies (Second Hit)
The exposed terminal GalNAc moiety on aberrant Gd-IgA1 is recognized by anti-glycan IgG antibodies, generating circulating immune complexes (ICs). Elevated Gd-IgA1 alone is not sufficient - a "second hit" (infection, drug, environmental trigger) is required to produce clinical disease.
Step 3: Immune Complex Deposition
These ICs deposit in the walls of small vessels in:
- Skin (dermal capillaries and venules)
- Glomerular mesangium (kidneys)
- Synovium (joints)
- Intestinal submucosa (gut)
Step 4: Complement Activation and Inflammation
IgA activates the alternative complement pathway, triggering neutrophil infiltration, release of proteolytic enzymes, and vessel wall damage - producing the characteristic leukocytoclastic vasculitis.
Additional Mediators
- Elevated TNF-alpha and IL-1beta
- Elevated vascular endothelial growth factor (VEGF) and endothelin
- Decreased factor XIII levels (associated with worse prognosis)
- High-titer IgA anti-endothelial cell antibodies (correlate with severe renal disease)
A high serum Ga-IgA1 level is not sufficient for the development of clinical symptoms. A "second hit" in the form of another environmental or inherited risk factor is required to produce IgA vasculitis.
- Firestein & Kelley's Textbook of Rheumatology
Key link to IgA nephropathy: The kidney histologic features of IgAV are indistinguishable from IgA nephropathy. Monozygotic twins developing IgAN and IgAV simultaneously have been documented. IgAV nephritis and IgAN share the same Gd-IgA1 pathogenic axis, differing primarily in systemic vs. isolated renal expression.
4. Triggers and Precipitants
- Upper respiratory tract infections (most common - streptococcal pharyngitis, viral URIs) - precede disease in up to 50% of cases
- Helicobacter pylori infection (implicated in childhood and adult cases)
- Drugs (antibiotics and others)
- Insect bites, foods, immunizations
- Underlying malignancy (important in adults - see below)
5. Clinical Features - The Classic Tetrad
A. Skin (100% of patients once fully established)
Palpable purpura is the hallmark:
- Starts as mottled purpura on extensor extremities
- Becomes hemorrhagic within ~1 day
- Begins to fade in ~5 days
- New crops appear over weeks
- Distributed on lower extremities, buttocks, and posterior legs (dependent areas)
- May include urticarial lesions, vesicles, necrotic purpura, or hemangioma-like lesions
- Koebnerization: streaky/linear purpura from pressure on inflamed vessels (see image below)
- Purpura above the waist may be a marker of renal involvement
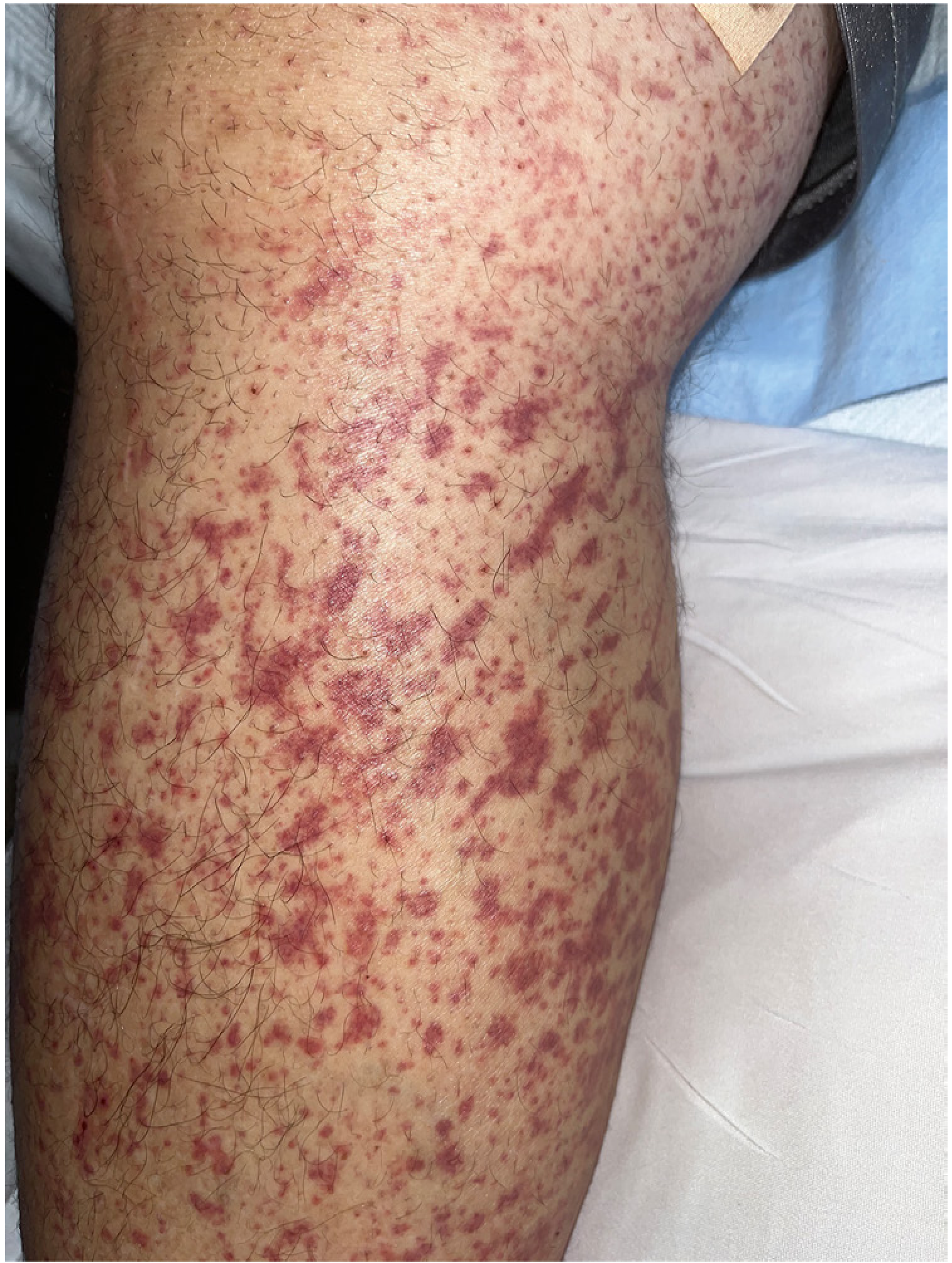
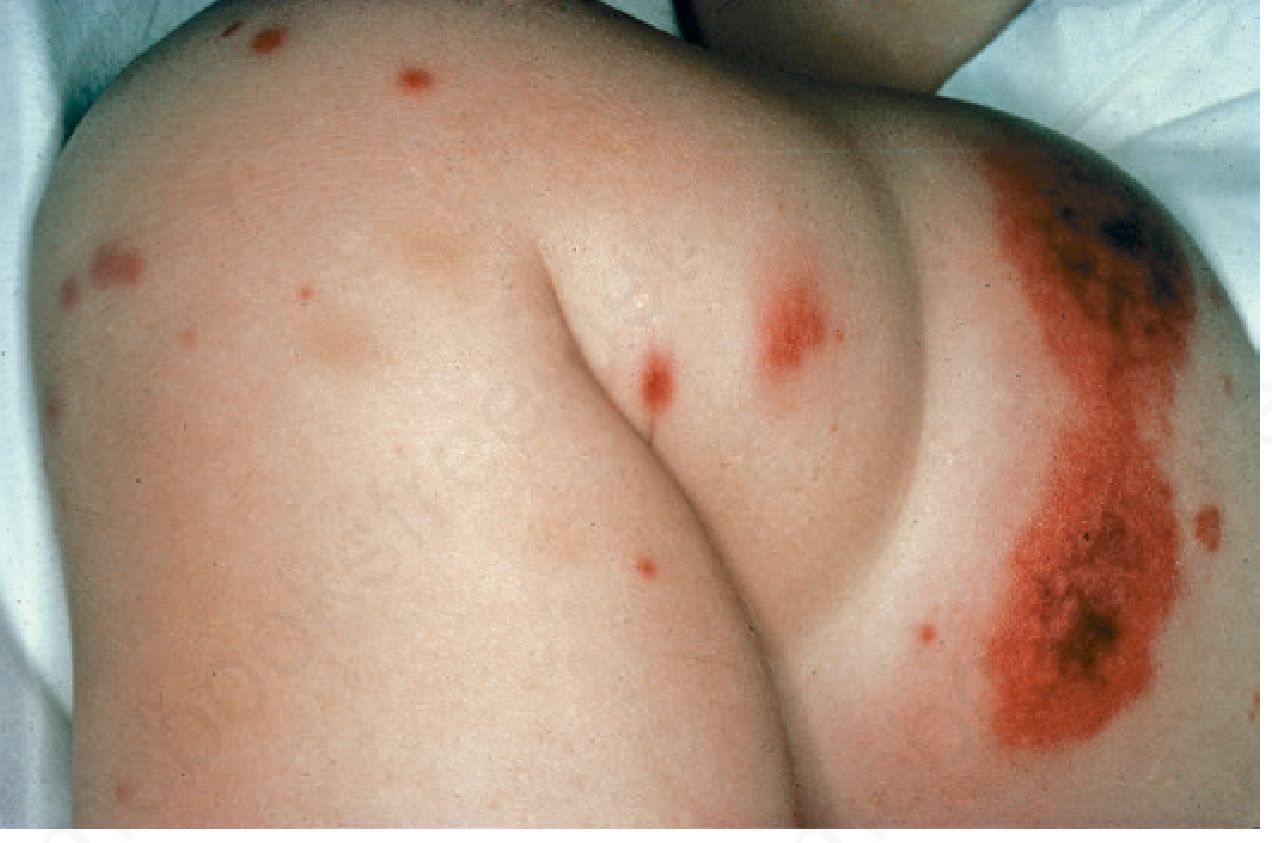
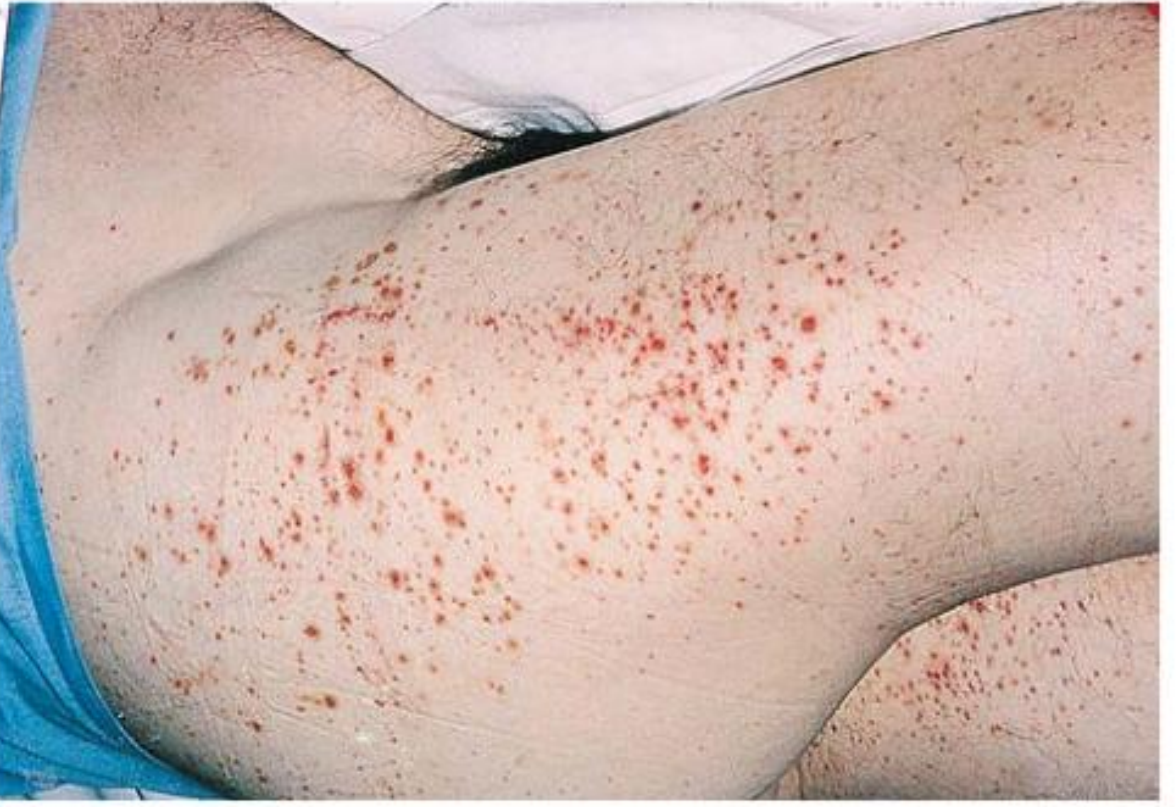
B. Joints (~63-82% of patients)
- Polyarthralgias progressing to arthritis
- Periarticular swelling around knees and ankles (most common joints)
- Usually without frank synovial effusion
- Non-destructive; resolves with the disease
C. Gastrointestinal Tract (~65-70% of patients)
- Colicky abdominal pain caused by gut vasculitis - typically occurs within 1 week after rash onset
- Nausea, vomiting, diarrhea, constipation
- Passage of blood and mucus per rectum (melena, hematochezia)
- Intussusception (classic risk - ultrasound required)
- Paralytic ileus, rebound tenderness, distension
- Endoscopy: purpura in upper or lower intestinal tract
- GI radiograph: "spiking" or "cobblestone" marbled appearance
- Important: GI symptoms may PRECEDE the rash in some cases, creating diagnostic difficulty and potentially leading to unnecessary exploratory surgery
D. Kidneys (10-50% in children; up to 70%+ in adults)
- Ranges from microscopic hematuria (+/- proteinuria) to nephrotic syndrome
- Red blood cell casts confirm glomerulonephritis
- Most cases are mild and self-limited in children
- Progressive glomerulonephritis and CKD/ESRD are uncommon in children but more prevalent in adults
- Nephrotic syndrome occurs in 20-30% of those biopsied
- AKI may develop from crescentic GN
6. Histopathology
Skin Biopsy (Light Microscopy)
Leukocytoclastic vasculitis of small vessels (venules/capillaries):
- Neutrophilic infiltration of vessel walls
- Nuclear dust (leukocytoclasis - fragmented neutrophil nuclei)
- Fibrinoid necrosis of vessel walls
- Extravasation of red blood cells
Skin Biopsy (Direct Immunofluorescence - DIF) - DIAGNOSTIC
Florid IgA deposition in dermal vessel walls, often with C3 and fibrin. This finding is pathognomonic - other forms of small vessel vasculitis may have trace IgA, but IgA is never the dominant immunoreactant in other diseases. Also IgM deposition in lesional skin may indicate renal involvement.
The "histamine trap test" (intradermal histamine injection, then biopsy at 4 hours) can identify IgA in vessels when skin lesions are absent but IgAV is suspected.
Renal Biopsy (Light Microscopy)
- Mesangial proliferative GN (most common)
- Diffuse endocapillary proliferation
- Crescentic GN (severe, rapidly progressive cases)
Renal Biopsy (Immunofluorescence)
- Diffuse mesangial IgA deposits (identical to IgA nephropathy)
- C3 and IgG co-deposition
- The MEST scoring system for IgAN may apply but its predictive value for IgAV nephritis has not been validated
7. Diagnosis
Clinical Diagnosis (especially in children)
In children with classic presentations, diagnosis is clinical without biopsy. The 2010 EULAR/PRINTO/PRES criteria require palpable purpura (mandatory) plus at least one of:
- Diffuse abdominal pain
- Any biopsy showing predominant IgA deposition
- Arthritis or arthralgia
- Renal involvement (hematuria and/or proteinuria)
1990 ACR Classification Criteria (for epidemiologic use)
At least 2 of 4:
- Age ≤20 years at disease onset
- Palpable purpura
- Acute abdominal pain
- Granulocytes on biopsy
Laboratory Findings
| Test | Finding |
|---|---|
| CBC | Mild leukocytosis; normal platelet count (key - distinguishes from ITP/TTP) |
| Serum IgA | Elevated in ~50% of patients |
| Complement (C3, C4) | Normal (unlike SLE, MPGN) |
| Coagulation studies | Generally normal |
| Urinalysis | Hematuria, RBC casts, proteinuria (if renal involvement) |
| BMP | BUN/Cr if renal involvement suspected |
| Ultrasound (abdomen) | Mandatory when abdominal pain/GI bleeding present - assess for intussusception |
Differential Diagnosis
- Septic purpura (meningococcemia) - thrombocytopenia present
- ITP / TTP - platelet count differentiates
- Streptococcal infection with rash + arthralgia
- Other systemic vasculitides (ANCA vasculitis, SLE vasculitis)
- Septic arthritis / reactive arthritis
- Acute surgical abdomen (when GI symptoms precede rash)
8. Special Population: Adults vs. Children
| Feature | Children | Adults |
|---|---|---|
| Course | Usually self-limited (6-16 wk) | More prolonged, recurrent |
| Renal severity | Usually mild, reversible | More frequent and severe |
| ESRD risk | 1-5% | Up to 11-40% (15-yr follow-up) |
| GI as first symptom | Less common | Less common as initial complaint |
| Malignancy association | Rare | Important - screen for solid tumors (NSCLC, prostate, renal), hematologic malignancies |
| Sex | Male predominance | 90%+ of malignancy-associated cases are male |
9. Prognostic Factors (Poor Prognosis)
- Nephrotic-range proteinuria (>1 g/day)
- Hypertension at presentation
- Crescents on renal biopsy
- Mesangial macrophages
- Tubulointerstitial fibrosis
- Decreased factor XIII levels
- Purpura above the waist
- Renal insufficiency at onset
- High-titer IgA anti-endothelial cell antibodies
10. Treatment
Supportive (all patients)
- Rest, hydration, pain management
- Acetaminophen for arthralgias
- NSAIDs for joint pain (use cautiously - avoid with severe renal involvement or active GI bleeding)
- Close follow-up: urinalysis + blood pressure monitoring for at least 6 months
Skin-Limited Disease
- Dapsone 50-200 mg/day
- Colchicine 0.6-1.2 mg twice daily
Gastrointestinal Disease
- H2 blockers for GI symptoms
- Prednisone 1 mg/kg/day (more effective for abdominal pain than analgesics; does NOT reduce renal complication risk)
- IVIG for persistent abdominal pain refractory to steroids
Renal Disease - A Spectrum of Interventions
The value of steroids for renal protection is controversial - randomized trials have not shown significant long-term benefit:
| Situation | Intervention |
|---|---|
| Mild hematuria/proteinuria | ACEi or ARB (target BP: 130/80 if proteinuria <1 g/day; 125/75 if >1 g/day) |
| Nephrotic syndrome | Prednisone + ACEi/ARB; treat as IgAN protocol |
| Crescentic nephritis | High-dose glucocorticoids + immunosuppressants (as for crescentic IgAN) |
| Severe/refractory renal disease | Mycophenolate mofetil (excellent option for immunosuppression + steroid-sparing) |
| Rapidly progressive GN | Cyclophosphamide + steroids; IVIG |
| Refractory skin + persistent GI | IVIG |
Cyclosporine A vs methylprednisolone for HSP nephritis has been studied in pediatric trials (Jauhola et al. 2011).
Despite this, it is prudent to treat aggressive renal involvement with an immunosuppressive regimen that includes high-dose glucocorticoids. Mycophenolate mofetil is an excellent option for severe renal disease.
- Firestein & Kelley's Textbook of Rheumatology
Malignancy-Associated IgAV
If a malignancy is identified, treating the primary tumor may lead to resolution of IgAV.
H. pylori-Associated IgAV (Adults with GI symptoms)
Test for H. pylori; successful eradication therapy may resolve the IgAV.
11. Emergency Department Approach (Rosen's Framework)
Indications for hospital admission:
- Cannot bear weight due to arthralgias
- Pain not controlled with oral medications
- Ongoing GI bleeding
- Renal involvement with fluid overload/AKI
- Severely elevated blood pressure
Critical ED workup:
- Urinalysis (mandatory)
- BMP if hematuria or hypertension
- Abdominal ultrasound if GI bleeding/abdominal pain (rule out intussusception)
- CBC (normal platelets excludes ITP)
12. Prognosis
- Overall prognosis is excellent, especially in children
- Mortality is exceedingly rare
- Most patients recover completely, often without therapy
- Illness duration: typically 6-16 weeks
- Children with gross hematuria usually do well
- Recurrent skin disease (multiple episodes over months) is not unusual but typically resolves over months to 1 year
- 1-5% of children progress to ESRD; in adults, up to 40% may develop CKD/ESRD at 15-year follow-up
- Persistent nephropathy (usually mild) in only ~8% of overall patients
13. Transplantation
For patients who progress to ESRD, kidney transplantation is performed. IgAV can recur in the transplant:
- ~50% of transplants: isolated IgA deposits in the graft
- Full-blown IgAN in the graft (clinically)
- Rarely: full systemic recurrence including rash
- Cadaveric donor may be preferable to living-related donor in children (controversial)
- Delaying transplantation after dialysis does not reduce recurrence risk
14. Recent Evidence (2024-2026)
Three recent systematic reviews are relevant:
-
Rituximab in IgAV nephritis (Xiong et al., 2024 - PMID 39249581): Evaluates the role of B-cell depletion for refractory nephritis - emerging option for severe/refractory IgAV nephritis.
-
Tacrolimus vs cyclophosphamide in HSP nephritis in children (Jiang et al., 2025 - PMID 40696685): Compares calcineurin inhibitor vs alkylating agent approaches in pediatric nephritis.
-
IgAV with necrotizing arteritis (Ribereau-Gayon et al., 2026 - PMID 42192510): A French multicenter study describing the severe subtype with medium-vessel necrotizing involvement - distinct clinical phenotype with worse prognosis.
Quick Reference Summary
HSP / IgA Vasculitis
├── Mechanism: Gd-IgA1 → anti-glycan IgG → ICs → vessel wall deposition
├── Trigger: URI, H. pylori, drugs, malignancy (adults)
├── Tetrad: Purpura + Arthralgias + Abdominal pain + Nephritis
├── DIF skin biopsy: Florid IgA deposition (DIAGNOSTIC)
├── Labs: Normal platelets, normal complement, elevated IgA (50%)
├── Kidney: Identical to IgAN on biopsy (MEST score)
├── Treatment:
│ ├── Mild: Supportive
│ ├── GI pain: Prednisone 1 mg/kg
│ ├── Renal: ACEi/ARB ± steroids ± MMF/CYC
│ └── Refractory: IVIG, rituximab (emerging)
└── Prognosis: Excellent in children; worse in adults (40% CKD at 15y)
Sources: Firestein & Kelley's Textbook of Rheumatology; Harrison's Principles of Internal Medicine 22E; Andrews' Diseases of the Skin; Comprehensive Clinical Nephrology 7E; Brenner and Rector's The Kidney; ROSEN's Emergency Medicine; National Kidney Foundation Primer on Kidney Diseases 8E
Give answer for viva neither too short nor too long
Reading File
Running Command
cd /home/daytona/workspace && pandoc --track-changes=all "/home/daytona/workspace/attachments/5baa4b4b-5aab-43d0-94bc-761e49bcc3f6/Exam questions ID4.docx" -o exam_questions.md 2>&1 && cat exam_questions.md
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
All 60 answers are complete. Here's a quick guide for using them in your viva:
Tips for this exam:
-
Repeated topics (PE appears 6 times, AF twice, AV blocks 4 times, Cardiomyopathies 3 times) - memorize those answers cold; the examiner will ask follow-up questions on them.
-
Overlapping answers - several questions share content (Q3 & Q5 = DCM; Q24 & Q26 & Q27 = Pericarditis; Q6 & Q37 & Q41 & Q54 = OSA; Q1 & Q8 & Q31 & Q34 = Scleroderma). Learn the full version once, then adapt.
-
Don't-miss points examiners love:
- Beck's triad + electrical alternans = tamponade
- PR depression on ECG = pericarditis (before even asking about ST elevation)
- Mobitz II → permanent pacemaker (unlike Mobitz I)
- Complete heart block → Stokes-Adams attacks + cannon a-waves
- SAAG ≥1.1 = portal hypertension ascites
- Anti-centromere = lcSSc/CREST; Anti-Scl-70 = dcSSc + ILD risk
- ACE inhibitors for scleroderma renal crisis = life-saving
- Lactulose + rifaximin for hepatic encephalopathy
This is a shared conversation. Sign in to Orris to start your own chat.