Investigations differentiating intravascular from extravascular hemolysis
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Searching Images
intravascular extravascular hemolysis comparison diagram
Reading File
Loading Image
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Here is a complete synthesis:
Investigations Differentiating Intravascular from Extravascular Hemolysis
Pathophysiologic Basis
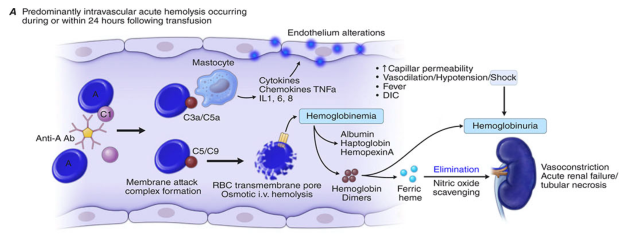
Intravascular hemolysis — RBCs lyse directly within the circulation (caused by complement fixation, mechanical injury, toxic/infectious agents). Free hemoglobin is released into plasma.
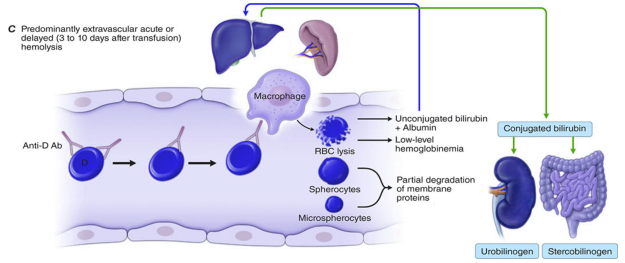
Extravascular hemolysis — RBCs are phagocytosed by macrophages in the spleen, liver, and bone marrow (the more common mechanism). Hemoglobin is metabolised intracellularly.
Comparison Table
| Investigation | Intravascular | Extravascular |
|---|---|---|
| Serum haptoglobin | Markedly ↓↓ (consumed by free Hb) | Normal to ↓ (may fall if severe) |
| Plasma/serum free Hb | ↑↑ (specific) | Normal / trace |
| Hemoglobinuria | Present (pink/red-brown urine) | Absent |
| Hemosiderinuria | Present (chronic/recurrent) | Absent |
| Methemalbumin | Present (if hemopexin depleted) | Absent |
| Hemopexin | ↓ (consumed) | Normal |
| Serum LDH | ↑↑ (more markedly) | ↑ |
| Serum indirect bilirubin | ↑ | ↑↑ (predominant finding) |
| Urine/fecal urobilinogen | ↑ | ↑↑ |
| Peripheral smear morphology | Schistocytes (fragmentation) | Microspherocytes (partial phagocytosis) |
| Splenomegaly | Absent | Often present |
| Reticulocyte count | ↑ | ↑ |
| Serum AST | Mildly ↑ | Mildly ↑ |
Key Differentiating Markers in Detail
1. Plasma Haptoglobin
- The most sensitive general hemolysis marker.
- Falls more steeply in intravascular hemolysis because free Hb floods the plasma and saturates haptoglobin (an α₂-globulin), forming Hb-haptoglobin complexes that are rapidly cleared by mononuclear phagocytes.
- In extravascular hemolysis, only small amounts of Hb escape from macrophages, so haptoglobin may be only mildly reduced.
- Caveat: haptoglobin is an acute-phase reactant — can be falsely normal in inflammation even with hemolysis. — Goldman-Cecil Medicine, p. 1674; Robbins Pathologic Basis of Disease, p. 596
2. Hemoglobinemia & Hemoglobinuria
- Specific for intravascular hemolysis.
- Free Hb in plasma imparts yellow → orange → pink color depending on level (>10 mg/dL = yellow; >25 mg/dL = pink).
- Once plasma Hb exceeds the renal tubular reabsorptive threshold, it spills into urine → hemoglobinuria (red-brown urine, dipstick positive for blood, no RBCs on microscopy).
- In extravascular hemolysis: no hemoglobinemia, no hemoglobinuria. — Henry's Clinical Diagnosis, p. 716
3. Hemosiderinuria
- Indicates chronic or recurrent intravascular hemolysis (e.g., PNH, mechanical heart valves).
- Mechanism: Hb filtered at the glomerulus is reabsorbed by proximal tubular cells → iron stored as hemosiderin → when tubular cells shed into urine, hemosiderin granules appear.
- Detected by Prussian blue stain of urine sediment (positive 3–4 days after an acute episode, persists for weeks).
- Absent in extravascular hemolysis. — Henry's Clinical Diagnosis, p. 716
4. Hemopexin & Methemalbumin
- When haptoglobin is exhausted, free Hb oxidises to methemoglobin → hemin groups are released and bound by hemopexin (a β-globulin) → cleared by liver.
- When hemopexin is also depleted, hemin binds to albumin → methemalbumin (detected by Schumm's test — spectroscopic).
- Both are specific markers of severe or sustained intravascular hemolysis. — Henry's Clinical Diagnosis, p. 716
5. LDH (Lactate Dehydrogenase)
- Released from lysed RBCs in both types, but more markedly elevated in intravascular hemolysis.
- LD₁ > LD₂ (reversal of normal isoenzyme pattern) — seen in hemolytic anemias.
- Upper normal ~207 IU/L; in hemolytic anemia may reach ~800 IU/L; in megaloblastic anemia (ineffective erythropoiesis), may reach several thousand. — Henry's Clinical Diagnosis, p. 716
6. Indirect (Unconjugated) Bilirubin & Urobilinogen
- Elevated in both types, but tend to predominate in extravascular hemolysis where haemoglobin is degraded intracellularly by macrophages.
- Bilirubin is unconjugated (bound to albumin) → does not appear in urine → no bilirubinuria (but urobilinogen is increased).
- Urine urobilinogen: normal 0.5–4 mg/24h; fecal urobilinogen: normal 40–280 mg/24h — both rise in hemolysis. — Henry's Clinical Diagnosis, p. 716
7. Peripheral Blood Smear Morphology
- Schistocytes (fragmented RBCs) → intravascular mechanical destruction (e.g., microangiopathic hemolytic anemia, prosthetic valves)
- Microspherocytes → partial membrane loss during extravascular phagocytosis (e.g., warm AIHA, hereditary spherocytosis)
- Note: Polychromasia and reticulocytosis are present in both. — Quick Compendium of Clinical Pathology, p. 8744
8. Splenomegaly
- Characteristic of extravascular hemolysis due to macrophage hyperplasia in the splenic cords.
- Absent in pure intravascular hemolysis. — Robbins Pathologic Basis of Disease, p. 596
Diagnostic Cascade Summary
General hemolysis markers (both types):
↑ LDH, ↓ haptoglobin, ↑ indirect bilirubin,
↑ reticulocytes, ↑ urobilinogen, polychromasia
↓
Intravascular-specific: Extravascular-specific:
• Hemoglobinemia • Splenomegaly
• Hemoglobinuria • Microspherocytes
• Hemosiderinuria • ↑↑ indirect bilirubin
• ↓↓ haptoglobin • ↑↑ urobilinogen/stercobilinogen
• Methemalbumin (Schumm +) • Normal plasma Hb
• ↓ hemopexin
• Schistocytes on smear
Illustrations (Harrison's Principles of Internal Medicine):
Intravascular hemolysis — complement-mediated RBC lysis releasing free Hb → hemoglobinemia, binding by haptoglobin/hemopexin, hemoglobinuria, nitric oxide scavenging:
Extravascular hemolysis — macrophage-mediated phagocytosis → spherocytes/microspherocytes → unconjugated bilirubin → urobilinogen/stercobilinogen:
Sources: Quick Compendium of Clinical Pathology, 5th ed. — Goldman-Cecil Medicine — Robbins & Cotran Pathologic Basis of Disease — Henry's Clinical Diagnosis and Management by Laboratory Methods — Harrison's Principles of Internal Medicine, 21st ed.
Causes
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
I have comprehensive data. Here is the full breakdown of causes:
Causes of Intravascular vs. Extravascular Hemolysis
Intravascular Hemolysis
RBCs are destroyed directly within the vasculature. The underlying mechanisms fall into four categories:
1. Mechanical / Physical Destruction
- Microangiopathic hemolytic anemia (MAHA) — fibrin strands or platelet thrombi shear RBCs:
- Thrombotic thrombocytopenic purpura (TTP)
- Hemolytic uremic syndrome (HUS — typical and atypical)
- Disseminated intravascular coagulation (DIC)
- HELLP syndrome
- Malignant hypertension
- Giant hemangioma / Kasabach-Merritt syndrome
- Prosthetic/dysfunctional heart valves (turbulent flow)
- March hemoglobinuria (foot-strike hemolysis — marathon runners, bongo drumming)
- Thermal injury (burns)
2. Complement Fixation on the RBC Surface
- ABO incompatible transfusion — IgM antibodies activate full complement cascade → MAC formation
- Paroxysmal nocturnal hemoglobinuria (PNH) — GPI-anchor deficiency → unregulated complement lysis (classic cause of chronic intravascular hemolysis)
- Paroxysmal cold hemoglobinuria (PCH) — Donath-Landsteiner IgG antibody fixes complement at cold temperatures, lysis occurs on rewarming
3. Infections
- Falciparum malaria ("blackwater fever") — direct RBC membrane rupture by merozoites + oxidant injury
- Babesiosis — intraerythrocytic parasite causing lysis
- Clostridial sepsis (C. perfringens) — phospholipases/lecithinases digest the RBC membrane
4. Toxic / Chemical
- Snake envenomation — phospholipases lyse membranes
- Spider bite (brown recluse — Loxosceles) — sphingomyelinase D
- Arsine gas (AsH₃) — severe oxidative Coombs-negative intravascular hemolysis + renal failure
- Copper (Wilson's disease crisis, ingestion)
- Lead poisoning (also inhibits erythropoiesis)
- Drug oxidants in G6PD deficiency — extreme oxidant load causes both intravascular and extravascular hemolysis
Extravascular Hemolysis
RBCs are phagocytosed by macrophages of the reticuloendothelial system — primarily the spleen, secondarily the liver and bone marrow. Triggers are anything that reduces RBC deformability or opsonises RBCs:
1. Membrane / Structural Defects (Congenital)
- Hereditary spherocytosis — spectrin/ankyrin/band 3 mutations → spherocytes trapped in splenic cords
- Hereditary elliptocytosis / pyropoikilocytosis
- Hereditary stomatocytosis
2. Enzyme Deficiencies
- G6PD deficiency — oxidant triggers (drugs: primaquine, dapsone; fava beans; infections) → Heinz body formation → extravascular removal (also some intravascular component with severe oxidant load)
- Pyruvate kinase (PK) deficiency — rigid, deformability-impaired RBCs
3. Hemoglobinopathies
- Sickle cell disease — deoxygenated HbS polymers → rigid sickled cells → splenic/hepatic trapping; also vaso-occlusion
- Thalassemia syndromes — excess unpaired globin chains precipitate → damaged membrane → phagocytosis; also ineffective erythropoiesis (intramedullary hemolysis)
- Other unstable hemoglobins
4. Immune-Mediated (Predominantly Extravascular)
- Warm autoimmune hemolytic anemia (WAIHA) — IgG-coated RBCs opsonised → splenic macrophage phagocytosis via FcγR (most common form of AIHA; occasional intravascular component at onset)
- Cold agglutinin disease (CAD) — IgM fixes C3b → primarily hepatic (Kupffer cell) extravascular clearance; massive complement activation can cause intravascular component
- Drug-induced immune hemolytic anemia — hapten, immune complex, or autoantibody mechanisms → extravascular (most) or intravascular
- Alloimmune — delayed hemolytic transfusion reactions (non-ABO; e.g., anti-Kidd/Jk^a)
- Hemolytic disease of the fetus and newborn (HDFN) — anti-D, anti-c, anti-Kell
5. Hypersplenism
- Congestive splenomegaly (portal hypertension, storage diseases, infiltrative disorders) — non-selective sequestration of all blood elements
6. Acquired Membrane Defects
- Spur cell anemia (acanthocytosis) — severe liver disease → abnormal membrane lipid composition → splenic trapping
- PNH — also causes extravascular component via C3b opsonisation (in addition to intravascular lysis)
Summary Mnemonic
| Intravascular | Extravascular | |
|---|---|---|
| Mechanism | Direct lysis in blood vessels | RES phagocytosis (spleen/liver) |
| Complement | Full cascade (MAC) | Opsonisation (C3b) only |
| Immune | ABO mismatch, PCH, PNH | WAIHA, CAD (mainly), HDFN |
| Mechanical | MAHA, prosthetic valves, march | — |
| Infection | Malaria, Babesia, Clostridium | Malaria (chronic mild) |
| Structural | — | HS, HE, PK deficiency |
| Hb defect | — | Sickle cell, thalassemia |
| Toxin | Arsine, snake venom, copper | — |
Sources: Quick Compendium of Clinical Pathology, 5th ed. — Goldman-Cecil Medicine — Robbins & Cotran Pathologic Basis of Disease — Robbins & Kumar Basic Pathology — Tietz Textbook of Laboratory Medicine, 7th ed.
Hdfn
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Searching Images
hemolytic disease fetus newborn HDFN pathophysiology
Loading Image
Reading File
I now have comprehensive coverage. Here is the full account of HDFN:
Hemolytic Disease of the Fetus and Newborn (HDFN)
Definition
HDFN (also called erythroblastosis fetalis) results from transplacental passage of maternal IgG alloantibodies directed against paternally inherited antigens on fetal red blood cells, causing hemolysis in the fetus and newborn.
Only IgG crosses the placenta (IgG1 and IgG3 most efficiently, IgG4 less so; IgM does not cross). — Quick Compendium of Clinical Pathology, 5th ed.
Pathophysiology
- Mother lacks a specific RBC antigen
- Fetal RBCs bearing that antigen enter maternal circulation (fetomaternal hemorrhage — FMH)
- Mother mounts an IgG alloimmune response
- IgG crosses the placenta → binds fetal RBC antigen
- IgG-coated fetal RBCs are destroyed by macrophages in the fetal spleen/liver → extravascular hemolysis
- Results in fetal anaemia → compensatory extramedullary erythropoiesis → erythroblastemia → if severe → hydrops fetalis
- After birth, neonatal jaundice develops (bilirubin cannot be cleared by the immature liver; unconjugated bilirubin crosses the blood-brain barrier → kernicterus)
Causes by Antibody System
1. Rh System — Most Clinically Significant
| Antibody | Features |
|---|---|
| Anti-D | Leading cause of severe HDFN; non-naturally occurring (requires prior exposure); usually absent in 1st pregnancy; accounts for majority of IUT-requiring cases |
| Anti-c | Second most severe; similar magnitude to anti-D; often from prior transfusion; >50% require IUT in some series |
| Anti-E | Usually mild; often found alongside anti-D (additive effect) |
| Anti-C, anti-e | Generally mild; rarely require IUT alone |
| Anti-G | Mimics anti-C + anti-D; high titer can cause significant fetal disease; RhIG indicated for invasive procedures |
Anti-D: Without RhIg, risk of sensitization in Rh− mother carrying Rh+ fetus ~15%. With antenatal + postnatal RhIg: 0.1%.
Sources of Rh(D) sensitization (FMH triggers):
- Normal pregnancy, delivery
- Spontaneous/elective abortion
- Chorionic villus sampling, amniocentesis, cordocentesis
- Placental abruption, ectopic pregnancy, trauma
2. Kell System
| Antibody | Features |
|---|---|
| Anti-K (K1) | Leading non-Rh cause; K1 antigen present in 9% Whites, 2% Blacks; causes hemolysis AND suppression of fetal erythropoiesis (unique mechanism — attacks erythroid precursors); critical titer = 4 (lower than other antibodies); 53% of Kell-alloimmunized pregnancies with K+ fetus required IUT in one series; majority from prior transfusion (blood not routinely Kell-typed) |
| Anti-k (K2) | Rare; can suppress erythropoiesis at low titers |
3. ABO Incompatibility — Most Common Overall
| Feature | Detail |
|---|---|
| Frequency | Most common cause of HDFN overall; ~20–25% of pregnancies at risk |
| Typical scenario | Group O mother with Group A or B fetus (O mothers have naturally occurring IgG anti-A,B) |
| Severity | Generally mild — clinically significant HDFN in only ~1% of at-risk pregnancies |
| Why 1st pregnancy affected | Anti-A,B in group O mothers are often already IgG — can cross placenta even in first pregnancy |
| Why milder than Rh | A/B antigens widely expressed on other tissues (antibody absorbed away); immature fetal antigen expression; lower density of A/B sites on fetal RBCs |
4. Other Blood Group Antibodies (selected)
| System | Antibody | Severity |
|---|---|---|
| Duffy | Anti-Fy^a | Neonatal jaundice; rarely severe |
| Kidd | Anti-Jk^a, anti-Jk^b | Mild HDFN |
| MNS | Anti-M (usually IgM) | Rarely causes HDFN; anti-M IgG rare |
| Colton | Anti-Co^a | Moderate |
| Diego | Anti-Wr^a | Moderate |
More than 60 different antibodies have been associated with HDFN. — Creasy & Resnik's Maternal-Fetal Medicine
Clinical Spectrum
| Severity | Features |
|---|---|
| Mild | Neonatal jaundice only; no significant anaemia |
| Moderate | Significant anaemia + jaundice; may need exchange transfusion |
| Severe | Severe fetal anaemia → high-output cardiac failure → generalised oedema (ascites, pleural/pericardial effusions, skin oedema) = hydrops fetalis |
| Kernicterus | Unconjugated bilirubin deposits in basal ganglia/brainstem → choreoathetosis, deafness, intellectual disability; preventable with phototherapy/exchange transfusion |
Investigations & Monitoring
Antenatal
| Test | Purpose |
|---|---|
| Antibody screen (indirect antiglobulin test) | At first prenatal visit in ALL pregnancies; repeat at 28 weeks in Rh− women |
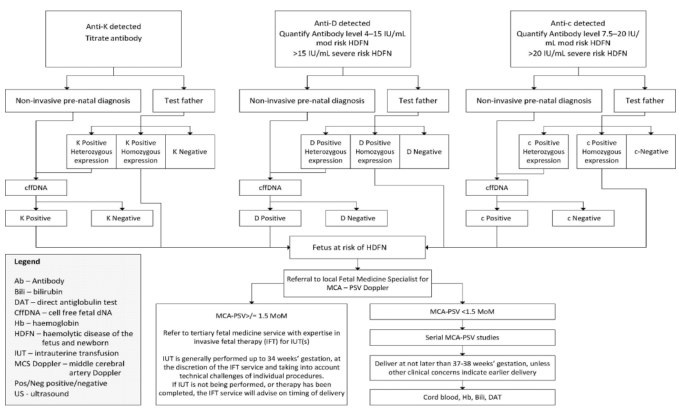
| Maternal antibody titer | Anti-D: critical titer = ≥16 → initiate fetal surveillance; Anti-K: critical titer = ≥4 |
| Anti-D quantification | 4–15 IU/mL = moderate risk; >15 IU/mL = severe risk |
| Anti-c quantification | 7.5–20 IU/mL = moderate risk; >20 IU/mL = severe risk |
| Paternal antigen typing | Determine whether fetus is at risk |
| Cell-free fetal DNA (cffDNA) | Non-invasive fetal genotyping from maternal blood → determines fetal antigen status (avoids IUT in antigen-negative fetus) |
| MCA peak systolic velocity (MCA-PSV) Doppler | Gold standard for non-invasive detection of fetal anaemia; >1.5 MoM (multiples of the median) → significant fetal anaemia → proceed to cordocentesis ± IUT |
| Amniotic fluid ΔOD₄₅₀ (Liley curve) | Older method; measures bilirubin in amniotic fluid; largely replaced by MCA Doppler |
| Cordocentesis (fetal blood sampling) | Confirms fetal Hb, blood group, DAT; performed when MCA-PSV >1.5 MoM |
Postnatal
| Test | Purpose |
|---|---|
| Cord blood Hb, bilirubin, DAT | At delivery in all at-risk neonates |
| Direct antiglobulin test (DAT) | Positive = IgG on neonatal RBCs (confirms immune hemolysis) |
| Serial serum bilirubin | Monitors rate of rise; guides phototherapy/exchange transfusion threshold |
Tests for Fetomaternal Hemorrhage (for RhIG dosing)
| Test | Details |
|---|---|
| Rosette test (qualitative) | Anti-D + D+ indicator cells form rosettes around D+ fetal cells; detects ≥10 mL fetal blood; if negative → standard 300 μg RhIg dose sufficient |
| Kleihauer-Betke test (quantitative) | Acid elution: HbF-containing (fetal) cells resist elution, stain pink; maternal cells appear as "ghosts"; determines % fetal cells → calculates volume of FMH |
| Flow cytometry | More sensitive and reproducible than Kleihauer-Betke for quantifying FMH |
RhIg dosing formula:
Dose (vials) = (Maternal blood volume in mL × proportion fetal cells) ÷ 30 Each 300 μg vial protects against 30 mL whole blood (15 mL packed RBCs) Always round up and add 1 vial; max 5 vials IM at one time
Management
Prevention (Rh HDFN)
- Rh− women without antibodies: RhIg 300 μg at 28 weeks + at delivery; also after any sensitising event (miscarriage, amniocentesis, trauma)
- Weak D type 1, 2, or 3: not at risk for alloimmunisation → RhIg not indicated (genotyping required to confirm)
- Already immunised (anti-D present): RhIg has no benefit
Fetal Treatment
- Intrauterine transfusion (IUT) — via cordocentesis; indicated when MCA-PSV >1.5 MoM (fetal Hb <70 g/L); generally performed up to 34 weeks' gestation
- Delivery at 37–38 weeks unless earlier delivery clinically indicated
Neonatal Treatment
- Phototherapy — converts unconjugated bilirubin to water-soluble isomers for excretion
- Exchange transfusion — removes IgG-coated RBCs, reduces bilirubin, corrects anaemia; antigen-negative, irradiated, CMV-safe blood used
- IVIG — reduces rate of haemolysis in severe cases
Management Flowchart
Sources: Quick Compendium of Clinical Pathology, 5th ed. — Creasy & Resnik's Maternal-Fetal Medicine — Henry's Clinical Diagnosis and Management by Laboratory Methods — Harriet Lane Handbook, 23rd ed. — Investigation and Management of Red Cell Antibodies in Pregnancy
What's the basis behind intra vs extravascular destruction
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Loading Image
Finding Sources
I now have everything needed from the immunology and blood bank texts. Here is the mechanistic basis:
Mechanistic Basis of Intravascular vs. Extravascular Destruction
The fundamental determinant is how far complement activation proceeds on the RBC surface — and whether the cell is lysed in the bloodstream or captured by phagocytes in the reticuloendothelial system (RES).
The Central Decision: Does Complement Go to Completion?
Antibody binds RBC antigen
↓
Classical pathway activated: C1 → C4 → C2 → C3
↓
C3b deposited on RBC membrane
↓
Two outcomes:
ARRESTED HERE PROCEEDS FURTHER
(Extravascular) (Intravascular)
↓ ↓
C3b → iC3b → C3dg C5 → C5b → C6,7,8,9
(by Factor I, Factor H, → MAC (C5b-6789)
CR1, DAF) → osmotic pore in
↓ membrane
Opsonised RBC → cell swells and bursts
phagocytosed by RES → intravascular lysis
Why Complement Usually Stops at C3 (Protecting Against Intravascular Lysis)
Normal RBCs express surface complement regulatory proteins that act as brakes:
| Protein | Gene | Mechanism |
|---|---|---|
| DAF (Decay Accelerating Factor, CD55) | GPI-anchored | Accelerates decay of C3 convertase (C4b2b); prevents more C3b deposition |
| CD59 (Protectin, MIRL) | GPI-anchored | Blocks the MAC assembly step (prevents C9 polymerisation) |
| CR1 (CD35) | Transmembrane | Binds C3b/C4b; cofactor for Factor I-mediated cleavage of C3b → iC3b |
| Factor I + Factor H | Plasma | Enzymatically cleave C3b → iC3b (no opsonin) → C3dg (inert) |
This is the precise defect in PNH: the GPI anchor biosynthesis gene (PIG-A) is mutated → CD55 and CD59 are absent → complement runs to completion → intravascular lysis. — Henry's Clinical Diagnosis and Management by Laboratory Methods
The Role of Immunoglobulin Class
The antibody class is the primary driver of which pathway predominates:
IgM → Intravascular Hemolysis
- IgM is a pentamer: 10 antigen-binding sites
- A single IgM molecule can simultaneously bind two Fc sites and activate C1q → classical pathway fires vigorously
- IgM fixes complement very efficiently → large amounts of C3b generated rapidly → overwhelms regulatory proteins → cascade proceeds to MAC
- Prototype: ABO incompatibility (anti-A, anti-B are IgM); cold agglutinin disease (IgM)
- IgM itself is too large to bind Fc receptors on macrophages → cannot directly cause extravascular phagocytosis
IgG → Predominantly Extravascular Hemolysis
- IgG is a monomer: two antigen-binding sites
- Requires multiple IgG molecules in close proximity to cooperatively activate C1q → complement activation is weak and slow
- Complement regulatory proteins (DAF, Factor I) stop the cascade at C3b → no MAC formed
- C3b-coated RBCs are opsonised and cleared by macrophages in the liver (via CR1/CR3 receptors for C3b/iC3b)
- Simultaneously, Fcγ receptors on splenic macrophages bind the IgG Fc tail directly → phagocytosis in the spleen
- Prototype: warm AIHA (IgG1, IgG3); HDFN due to anti-D
Exception: IgG anti-Kidd antibodies are highly complement-fixing and can cause intravascular hemolysis despite being IgG. This is because anti-Kidd fixes complement unusually efficiently. — Henry's Clinical Diagnosis and Management by Laboratory Methods
Mechanism of Extravascular Destruction in Detail
Step 1 — Opsonisation
IgG Fc tail and/or C3b on the RBC surface act as opsonins.
Step 2 — Capture in splenic cords / hepatic sinusoids
- Splenic macrophages: bind IgG via FcγRI (CD64) and FcγRIII (CD16)
- Hepatic Kupffer cells: bind C3b via CR1 and iC3b via CR3 (CD11b/CD18)
Step 3a — Complete phagocytosis
The macrophage engulfs the entire RBC → haemoglobin metabolised intracellularly → bilirubin + CO released
Step 3b — Partial phagocytosis → microspherocyte formation
The macrophage takes a "bite" of membrane from the opsonised RBC → RBC membrane area decreases but volume stays the same → RBC becomes a microspherocyte → reduced deformability → trapped in splenic cords on subsequent passes → eventually destroyed
RBCs coated with both IgG and C3b are cleared faster via the liver; those coated with IgG alone are destroyed more slowly in the spleen. — Henry's Clinical Diagnosis and Management by Laboratory Methods
Non-Immune Mechanisms
The same principle applies to non-immune causes:
| Mechanism | Basis | Type |
|---|---|---|
| Mechanical shear (MAHA, valves) | Physical fragmentation of RBC membrane by fibrin strands or turbulence | Intravascular |
| Reduced deformability (HS, spherocytosis, G6PD Heinz bodies) | RBCs cannot negotiate the 3 μm splenic sinus aperture → trapped and phagocytosed | Extravascular |
| Direct membrane toxins (Clostridium lecithinases, snake phospholipases) | Enzymatic destruction of phospholipid bilayer | Intravascular |
| Intracellular parasites (malaria, babesiosis) | Rupture of RBC on merozoite egress | Intravascular |
Summary Diagram
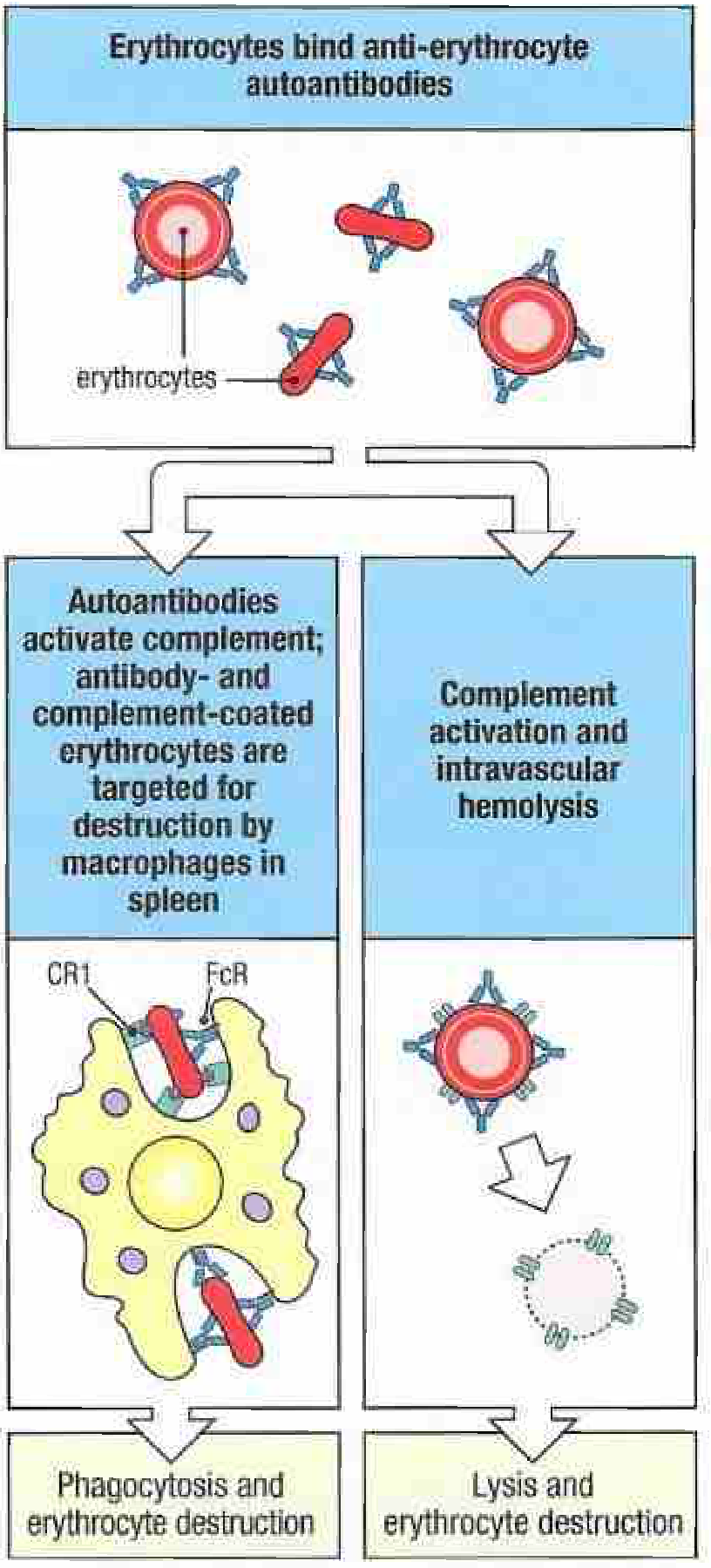
Left: IgG-coated RBCs bound by FcR and CR1 on splenic macrophages → phagocytosis and extravascular destruction. Right: Efficient complement activation to MAC → osmotic lysis within the vasculature. — Janeway's Immunobiology, 10th ed.
One-Line Summary
Intravascular hemolysis occurs when complement activation is rapid and overwhelming (typically IgM, or complement-fixing IgG, or direct toxic/mechanical injury), bypassing regulatory proteins and forming the MAC. Extravascular hemolysis occurs when complement activation stalls at C3b (or antibody is IgG without complement), leaving RBCs opsonised for phagocytosis by splenic and hepatic macrophages via Fcγ and complement receptors.
Sources: Henry's Clinical Diagnosis and Management by Laboratory Methods — Janeway's Immunobiology, 10th ed. — Robbins & Cotran Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.