Thalassemia pediatrics
Reading File
Finding Sources
Searching PubMed
thalassemia children management
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
I now have thorough, authoritative content from Harrison's (2025), Goldman-Cecil, and Robbins. Let me compile the comprehensive answer.
Thalassemia in Pediatrics
Definition and Genetics
Thalassemias are inherited disorders caused by mutations in globin genes that decrease synthesis of alpha- or beta-globin chains. The resulting imbalance leads to:
- Deficiency of functional hemoglobin (HbA) → hypochromic, microcytic red cells
- Excess unpaired globin chains that precipitate within erythroid precursors → membrane damage, ineffective erythropoiesis, and hemolysis
-
Beta-globin gene: chromosome 11 (single gene per haploid genome)
-
Alpha-globin genes: chromosome 16 (two genes per haploid genome, four total)
-
Autosomal codominant inheritance
-
Endemic in Mediterranean, Middle East, tropical Africa, Indian subcontinent, and Southeast Asia - consistent with a heterozygote protective effect against falciparum malaria
-
Robbins & Kumar Basic Pathology, p. 602
-
Harrison's Principles of Internal Medicine 22E, p. 820
Classification
Beta-Thalassemia
Caused by point mutations (most common) or small deletions affecting:
- Splicing mutations - most common cause of beta+ thalassemia
- Promoter region mutations - reduce transcription by 75-80%; cause beta+ thalassemia
- Chain terminator / frameshift mutations - most common cause of beta0 thalassemia (no beta-globin produced)
| Clinical Syndrome | Genotype | Hb (g/dL) / MCV (fL) | Hb Fractions | Clinical Features |
|---|---|---|---|---|
| Beta-thalassemia trait (minor) | Heterozygous | 10-14 / 60-80 | HbA2 elevated (4-6%), HbF mildly elevated | Asymptomatic; mild or no anemia; red cell abnormalities on smear |
| Non-transfusion-dependent (intermedia) | Variable | 7-12 / 65-80 | HbA 60-90%, HbF 10-40% | Moderately severe anemia; no regular transfusions required; iron loading, thromboembolism, pulmonary HTN are key risks |
| Transfusion-dependent (major / Cooley's) | Homozygous or compound heterozygous | 2-4 / 50-80 | HbF 90-100%, HbA 0-5% | Severe anemia requiring regular transfusion; iron chelation required |
| HbE-beta thalassemia | HbE + beta mutation | 5-8 / 60-70 | HbE 50-70%, HbF 30-50% | Common in Southeast Asia; may be most prevalent severe thalassemia globally; transfusion dependence depends on the beta mutation |
Harrison's Principles of Internal Medicine 22E, Table 103-4
Alpha-Thalassemia
Caused almost exclusively by gene deletions (unlike beta-thalassemia).
| Syndrome | Gene Deletions (of 4) | Features |
|---|---|---|
| Silent carrier | 1 (-alpha/alpha alpha) | Normal; no clinical features |
| Alpha-thalassemia trait | 2 (--/alpha alpha or -alpha/-alpha) | Microcytosis, mild anemia |
| HbH disease | 3 (--/-alpha) | Hb 8-9 g/dL; moderate hemolytic anemia; splenomegaly; no regular transfusions needed |
| Hydrops fetalis | 4 (--/--) | Incompatible with life without intrauterine transfusions; severe fetal anemia, heart failure |
Robbins, Cotran & Kumar Pathologic Basis of Disease
Pathophysiology
In beta-thalassemia, the primary mechanism is ineffective erythropoiesis:
- Excess alpha-chains precipitate in erythroid precursors
- Inclusions cause membrane lipid oxidation and damage
- Most precursors are destroyed in the bone marrow before reaching circulation
- This drives:
- Massive bone marrow expansion ("chipmunk" facies, "hair on end" skull X-ray, pathologic fractures)
- Hepatosplenomegaly (extramedullary hematopoiesis + hemolysis)
- Increased intestinal iron absorption
- Iron overload in liver, heart, and endocrine organs (even without transfusions, but vastly worsened by them)
In poorly treated beta-thalassemia major, children develop:
- Severe growth retardation
- Frontal bossing, maxillary overgrowth
- Hepatosplenomegaly
- Pathologic bone fractures
- Progressive cardiopulmonary failure
Harrison's Principles of Internal Medicine 22E, p. 820
Clinical Features in Children
Presentation
- Beta-thalassemia major typically presents at 6-24 months of age, when HbF production physiologically declines and the switch to beta-globin synthesis is expected
- Progressive pallor, failure to thrive, abdominal distension from hepatosplenomegaly
- Jaundice (hemolytic)
- Characteristic facies from bone marrow expansion if undertreated
Physical findings
- Pallor, jaundice, scleral icterus
- Massive splenomegaly (may also enlarge liver)
- Bony deformities: prominent malar eminences, frontal bossing, "hair on end" calvarium on skull X-ray
- Growth retardation
Diagnosis
Laboratory
- CBC: severe microcytic, hypochromic anemia; Hb 2-4 g/dL in thalassemia major
- Peripheral smear: target cells, hypochromic cells, nucleated red cells, basophilic stippling
- Elevated reticulocytes; elevated bilirubin (indirect)
- Elevated serum ferritin and iron
- HPLC (high-performance liquid chromatography): elevated HbF (90-100%), absent or minimal HbA, elevated or normal HbA2 in beta-thalassemia major
Genetic testing
- Mutation analysis of beta-globin gene is essential before genetic counseling and antenatal diagnosis
- Sequencing identifies the specific mutation(s) - critical for predicting phenotype severity
Heterozygotes (thalassemia minor/trait)
- Mild microcytic anemia or normal Hb
- Elevated HbA2 (>3.5%) on HPLC is the key diagnostic finding
- Must be distinguished from iron deficiency anemia (which lowers HbA2)
Harrison's Principles of Internal Medicine 22E, p. 820-821
Management
1. Regular Blood Transfusions (Transfusion-Dependent Thalassemia)
- Goal: maintain pre-transfusion Hb >9-10.5 g/dL to suppress ineffective erythropoiesis, allow normal growth and development, and prevent bone changes
- Frequency: every 2-5 weeks
- Use leukoreduced packed red cells to minimize alloimmunization and transfusion reactions
- Well-transfused children have relatively normal growth and can enter puberty
- Decision to initiate lifelong transfusion: based on definitive diagnosis, severity of anemia on repeated measurement, degree of ineffective erythropoiesis, and clinical criteria (failure to thrive, bone changes)
Goldman-Cecil Medicine, p. 219
2. Iron Chelation Therapy
Iron overload is the primary cause of morbidity and mortality in transfusion-dependent thalassemia. Each unit of packed red cells contains ~200-250 mg of iron, and the body has no mechanism to excrete excess iron.
- Initiate when serum ferritin >1000 ng/mL or after ~10-20 transfusions, typically by age 2-3 years
- Monitor iron burden with liver MRI (R2 or T2)** and cardiac MRI (T2)* - critical for guiding chelation intensity
Chelating agents:
| Agent | Route | Notes |
|---|---|---|
| Deferoxamine (desferrioxamine, DFO) | SC/IV infusion (8-12 hrs, 5-7 nights/week) | Most established; highly effective; compliance is major barrier in children |
| Deferasirox (Exjade, Jadenu) | Oral (once daily) | Preferred for children; renal monitoring required |
| Deferiprone (Ferriprox) | Oral (3x daily) | Especially effective for cardiac iron; agranulocytosis risk (CBC monitoring weekly); used in combination therapy |
- Cardiac iron (assessed by T2* MRI) is the key predictor of heart failure and arrhythmia - the leading cause of death in undertreated patients
Goldman-Cecil Medicine; Miller's Anesthesia 10e
3. Splenectomy
- Considered when annual blood consumption increases progressively causing significant increase in iron stores despite good chelation, or in the presence of symptomatic hypersplenism
- Post-splenectomy infection is a major risk (especially encapsulated organisms: Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis)
- Vaccinate against all encapsulated organisms prior to splenectomy
- Prophylactic penicillin post-splenectomy (particularly in young children)
- Splenectomy may be delayed or avoided with optimal transfusion management
4. Hematopoietic Stem Cell Transplantation (HSCT)
- The traditional curative option
- Matched sibling donor HSCT is curative in >80% of cases
- Best results in youngest, well-chelated patients with fewer prior transfusions
- Only ~1/3 of patients have matched sibling donors
- Risks: graft failure, graft rejection, graft-versus-host disease (GVHD), transplant-related mortality 5-20%
- Haploidentical and unrelated donor results are improving
Harrison's Principles of Internal Medicine 22E, p. 821
5. Gene Therapy (Approved, Cutting-Edge)
Two approved approaches for transfusion-dependent beta-thalassemia:
- CRISPR/Cas9 editing (Casgevy - exa-cel): Downregulates BCL11A to reactivate HbF production. Results: Hb ≥12 g/dL and HbF ≥10 g/dL, leading to transfusion independence in >90% of patients aged 12-35
- Lentiviral gene addition (betibeglogene autotemcel - beti-cel): Inserts a functional beta-globin gene (HbA^T87Q). Best results in non-beta0/beta0 genotypes, though higher viral titers have improved outcomes in beta0/beta0
Harrison's Principles of Internal Medicine 22E (2025)
6. Adjuncts
- Folic acid supplementation (chronic hemolysis depletes folate)
- Luspatercept (erythroid maturation agent): reduces transfusion requirements in adults; evidence is expanding
- Hydroxyurea: beneficial in some non-transfusion-dependent patients and HbE-beta thalassemia by raising HbF
Complications in Children
| Complication | Notes |
|---|---|
| Growth retardation | Often from delayed/inadequate transfusions, but can occur even in well-transfused children |
| Delayed puberty (50%) | Hypothalamic-pituitary iron deposition; hypogonadism |
| Cardiac disease | Leading cause of death; chronic anemia + iron toxicity + pulmonary HTN; monitor with cardiac MRI T2* |
| Hepatic disease | Fibrosis → cirrhosis from iron overload + hepatitis (transfusion-transmitted) |
| Endocrinopathies | Diabetes mellitus, hypothyroidism, hypoparathyroidism, adrenal insufficiency |
| Bone disease | Osteoporosis in ~50%; pathologic fractures from marrow expansion; extramedullary masses in thalassemia intermedia |
| Infections | Transfusion-associated (hepatitis B, C, HIV historically); post-splenectomy sepsis; Yersinia with iron overload |
| Thromboembolism | Multifactorial; platelet activation, red cell-endothelial interactions; higher risk post-splenectomy and in thalassemia intermedia |
Harrison's Principles of Internal Medicine 22E, Table 103-5
HbH Disease in Children
- HbH patients have Hb ~8-9 g/dL and generally do not need regular transfusions
- Folic acid 2-5 mg/day is recommended, especially in pediatric patients
- Hemolytic crises may occur during acute infections - require prompt blood transfusion and infection treatment
- Avoid oxidant drugs
Goldman-Cecil Medicine
Thalassemia Minor (Trait) in Children
- No specific treatment required
- Genetic counseling for parents is essential
- During pregnancy (for carrier mothers): monitor during 2nd and 3rd trimesters; folic acid support
- If true iron deficiency coexists: iron supplementation with monitoring of ferritin and transferrin saturation
- Do not administer empiric iron to children with thalassemia trait presenting with microcytic anemia (will worsen iron overload)
Key Points Summary
- Thalassemia major presents in infancy/early childhood; diagnosis confirmed by HPLC showing markedly elevated HbF
- Treatment foundation: regular transfusions (Hb target >9-10.5 g/dL) + iron chelation (typically starting age 2-3 years)
- Cardiac iron overload (T2* MRI) is the most feared complication - the leading cause of death
- HSCT from a matched sibling offers cure in >80%; best outcomes in youngest, well-chelated patients
- Gene therapy (CRISPR-based and lentiviral) is now approved and achieves transfusion independence in >90% of eligible patients
- Post-splenectomy: lifelong penicillin prophylaxis and vaccinations against encapsulated organisms
Sources:
- Harrison's Principles of Internal Medicine, 22E (2025), Chapter 103
- Goldman-Cecil Medicine, International Edition
- Robbins & Kumar Basic Pathology
- Robbins, Cotran & Kumar Pathologic Basis of Disease
SAM pediatrics
Reading File
Finding Sources
Searching PubMed
severe acute malnutrition children management
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
I now have comprehensive textbook content and clinical images. I have enough to write a thorough, well-structured answer on SAM in pediatrics.
Severe Acute Malnutrition (SAM) in Pediatrics
Definition
The WHO defines SAM as a life-threatening condition characterized by any one of the following:
| Diagnostic Criterion | Threshold |
|---|---|
| Weight-for-height Z-score (WHZ) | <-3 SD below WHO median |
| Mid-upper arm circumference (MUAC) | <115 mm (6-59 months) |
| Bilateral pitting edema of lower limbs | Present (nutritional) |
Children with SAM are 9 times more likely to die than well-nourished children. In 2022, approximately 13.6 million children under 5 years had SAM globally (2.1% of all under-fives), with >75% living in Asia and 22% in Africa.
Robbins & Kumar Basic Pathology; Park's Textbook of Preventive and Social Medicine
Classification: The SAM Spectrum
SAM was previously called Protein-Energy Malnutrition (PEM). It ranges across a spectrum with Marasmus and Kwashiorkor at opposite ends.
Two key protein compartments help understand the distinction:
- Somatic compartment: skeletal muscle proteins - depleted more in marasmus
- Visceral compartment: liver and organ proteins (albumin, transferrin) - depleted more in kwashiorkor
1. Marasmus ("Dry" SAM)
Cause: Severe deficiency of both calories and protein over months to years.
Pathophysiology:
- Body catabolizes somatic (muscle) protein for energy
- Visceral compartment relatively preserved - serum albumin normal or near-normal
- Subcutaneous fat mobilized as fuel
- Low leptin stimulates hypothalamic-pituitary-adrenal axis → high cortisol → lipolysis
Clinical features:
- Weight <60% of expected for age/height/sex
- Severe muscle wasting and loss of subcutaneous fat
- Emaciated extremities - "old man face" with head appearing disproportionately large for body
- No edema
- Dry, thin, pale, lax, and wrinkled skin
- Fine lanugo-like hair; thin, slow-growing hair that falls out
- Anemia (multivitamin deficiency)
- Immune deficiency (T-cell-mediated) → recurrent infections
- Growth retardation
2. Kwashiorkor ("Wet" or Edematous SAM)
Cause: Protein deprivation relatively greater than caloric reduction - a low protein/energy ratio diet. Classic setting: child weaned early and fed predominantly carbohydrate diet. (Kwashiorkor in Ga language of Ghana = "the sickness the older child gets when the new baby is born.")
Common in impoverished areas of Africa, Southeast Asia, and Central America.
Pathophysiology:
- Visceral protein compartment severely depleted → hypoalbuminemia
- Hypoalbuminemia → reduced plasma oncotic pressure → generalized/dependent edema
- Reduced synthesis of lipoprotein carrier proteins → fat cannot be exported from liver → fatty liver
- True weight loss masked by fluid retention; weight typically 60-80% of expected
Clinical features:
- Bilateral pitting edema (puffiness of face, hands, and legs; ascites)
- Relatively preserved subcutaneous fat and muscle (masked by edema)
- Hepatomegaly (fatty liver)
- Characteristic skin lesions: alternating zones of hyperpigmentation, desquamation, and hypopigmentation ("flaky paint" dermatosis)
- Hair changes: loss of color, alternating bands of pale and darker color ("flag sign" / signe de la bandera), straightening, fine brittle texture, easy hair pull
- Apathy, listlessness, irritability, loss of appetite
- Small bowel mucosal atrophy → disaccharidase deficiency → may not tolerate full-strength milk-based feeds initially
- Immune deficiency → secondary infections
- Anemia (multifactorial: iron, folate, protein deficiency + chronic infection)
3. Marasmic Kwashiorkor
Features of both - the most common overlap presentation in clinical practice.
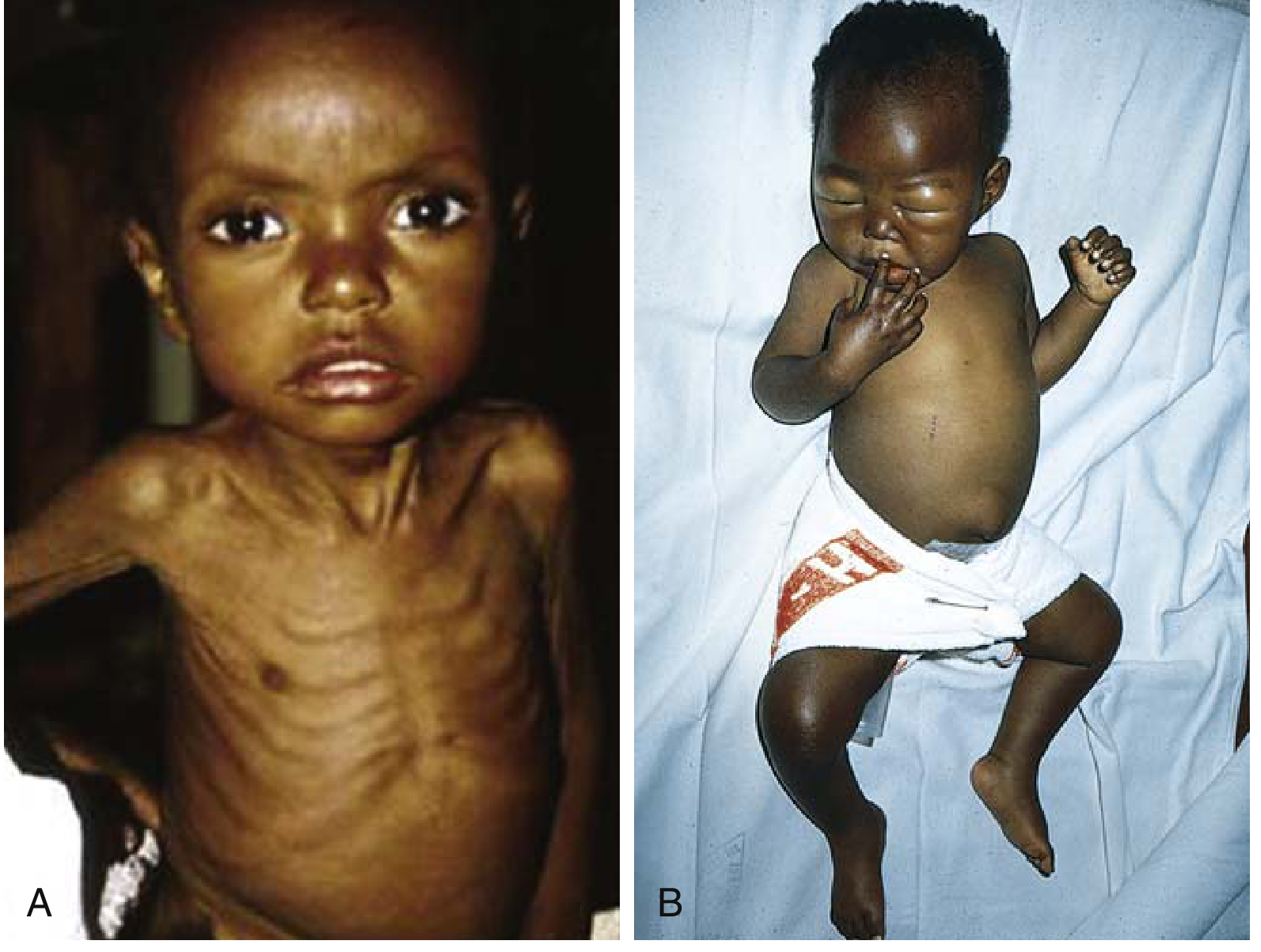
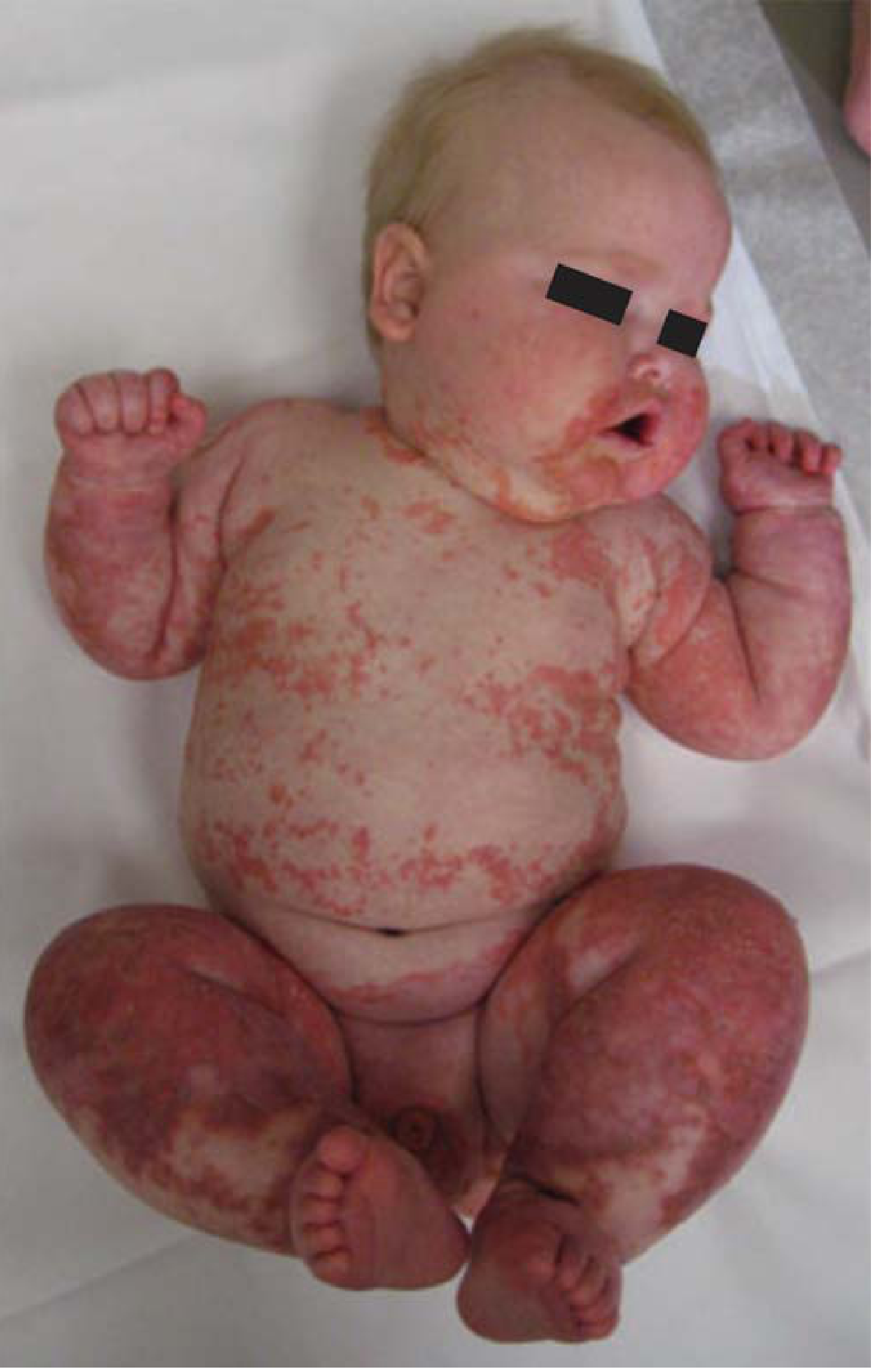
Robbins & Kumar Basic Pathology, pp. 289-291
Marasmus vs. Kwashiorkor - Comparison
| Feature | Marasmus | Kwashiorkor |
|---|---|---|
| Primary deficit | Calories + protein (total) | Protein >> Calories |
| Protein compartment affected | Somatic (muscle) | Visceral (liver/albumin) |
| Serum albumin | Normal or mildly low | Markedly low |
| Edema | Absent | Present (bilateral pitting) |
| Weight | <60% expected | 60-80% expected (masked by edema) |
| Subcutaneous fat | Severely depleted | Relatively preserved |
| Muscle wasting | Severe | Moderate |
| Liver | Normal | Enlarged and fatty |
| Skin | Dry, lax, wrinkled | Flaky paint dermatosis |
| Hair | Thin, falls out | Flag sign, depigmented, brittle |
| Behavior | Anxious, alert | Apathetic, listless |
| Gut mucosal atrophy | Minimal | Present → lactose intolerance |
Pathology (Morphology)
- Growth failure (universal)
- Peripheral edema in kwashiorkor
- Loss of body fat and muscle atrophy (more marked in marasmus)
- Liver: enlarged and fatty in kwashiorkor (not marasmus); superimposed cirrhosis is rare
- Small bowel: decreased mitotic cells, mucosal atrophy, loss of villi and microvilli in kwashiorkor → disaccharidase deficiency (especially lactase)
- Bone marrow: hypoplastic with decreased red cell precursors
- Thymus and lymphoid tissue: atrophy (more in kwashiorkor)
- Brain (in infants with early-onset SAM): cerebral atrophy, reduced neurons, impaired myelination
Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 413
Early Detection and Screening
| Tool | Normal | Mild-Moderate Malnutrition | Severe Malnutrition |
|---|---|---|---|
| MUAC (1-5 years) | >13.5 cm | 12.5-13.5 cm | <12.5 cm (SAM: <11.5 cm) |
| WHZ | >-2 SD | -2 to -3 SD | <-3 SD |
| Weight-for-age | >-2 SD | -2 to -3 SD | <-3 SD (underweight) |
- Growth charts (weight monitoring) - the most practical field tool
- Skinfold thickness (subcutaneous fat)
- Mid-arm circumference (somatic protein reserve)
- Serum albumin and transferrin (visceral protein reserve)
Management: WHO 10-Step Protocol
SAM management is divided into two phases and follows the WHO 10-step approach. Children with SAM + medical complications require inpatient care; uncomplicated SAM may be managed through outpatient/community-based programs.
Triage: Who Needs Inpatient Admission?
Inpatient care indicated if any of:
- Severe edema (grade 3) or edema with complications
- Severe anorexia (fails appetite test)
- Altered consciousness / convulsions
- Severe dehydration
- Respiratory distress / pneumonia
- Fever >39°C or hypothermia <35.5°C
- Severe anemia or signs of shock
- Hypoglycemia
Phase 1 - Stabilization (Days 1-7): Treat Life-Threatening Conditions
Step 1: Treat/Prevent Hypoglycemia
- Blood glucose <3 mmol/L (<54 mg/dL) = emergency
- Give 50 mL of 10% dextrose or 10% sucrose orally/NGT, OR 5 mL/kg 10% dextrose IV
- Start feeds immediately every 2-3 hours (F-75)
- Never fast a SAM child
Step 2: Treat/Prevent Hypothermia
- Temperature <35.5°C rectally or <36°C axillary
- Warm the child (skin-to-skin, blankets, warm room); avoid draughts
- Feed every 2 hours (generates heat)
Step 3: Treat/Prevent Dehydration
- SAM children are often dehydrated but IV fluids are dangerous (risk of cardiac overload)
- Use ReSoMal (Rehydration Solution for Malnutrition) - lower sodium, higher potassium and glucose than standard ORS
- 5 mL/kg every 30 minutes for 2 hours (oral/NGT), then 5-10 mL/kg/hr alternating with F-75 for up to 10 hours
- IV fluid ONLY for severe shock or cholera; use Ringer's lactate + 5% dextrose at 15 mL/kg/hr
Step 4: Correct Electrolyte Imbalances
- SAM children have excess body sodium but deficient intracellular potassium and magnesium
- Use electrolyte/mineral solution (potassium + magnesium + zinc) added to feeds
- Do NOT give diuretics for edema (dangerous - electrolyte depletion)
- Do NOT give iron during stabilization phase (potentiates infections, generates free radicals)
Step 5: Treat Infections
- Even without obvious signs of infection, assume infection exists in SAM
- Routine antibiotic therapy: amoxicillin (or ampicillin + gentamicin if severely ill) for all SAM children
- Treat specific infections as identified
- Immunize against measles (if not done) once condition stable (but not in shock)
Step 6: Correct Micronutrient Deficiencies
- Give daily for at least 2 weeks:
- Folic acid (5 mg on day 1, then 1 mg/day)
- Zinc (2 mg/kg/day)
- Copper (0.3 mg/kg/day)
- Multivitamins
- Vitamin A (only if signs of deficiency or recent measles): 200,000 IU on days 1, 2, and 14
- Do NOT give iron in Phase 1 - wait until Phase 2 (rehabilitation)
Step 7: Start Cautious Feeding (F-75)
- F-75 therapeutic milk: 75 kcal/100 mL, 0.9 g protein/100 mL (low protein, low sodium, low osmolarity)
- Initial volume: 100 mL/kg/day divided every 2-3 hours (8-12 feeds/day)
- Purpose: repair metabolic processes without overwhelming the gut or heart; not for weight gain yet
- If child cannot eat: give by nasogastric tube
Phase 2 - Rehabilitation (Weeks 2-6+): Intensive Feeding and Catch-up Growth
Step 8: Achieve Catch-up Growth
- Transition to F-100 (100 kcal/100 mL, 2.9 g protein/100 mL) or RUTF (Ready-to-Use Therapeutic Food)
- RUTF (e.g., Plumpy'Nut): peanut-paste-based, 500 kcal/92g sachet, lipid-based, does not require refrigeration, can be given at home
- Volume increases stepwise to 150-220 mL/kg/day
- Target weight gain: >10 g/kg/day indicates successful catch-up
- Now add iron: 3 mg/kg/day elemental iron
- Stimulation: emotional and physical (structured play, sensory stimulation)
- Continue antibiotics if infection ongoing
Step 9: Provide Sensory Stimulation and Emotional Support
- Cheerful, stimulating environment
- Structured play therapy 15-30 min/day
- Tender loving care; involve mother/caregiver
Step 10: Prepare for Follow-up After Recovery
Discharge Criteria
- MUAC ≥12.5 cm (or WHZ ≥-2 SD)
- No edema for at least 2 weeks
- Eating well, alert and active
- No acute medical issues
- Caregiver education complete
Discharge does not mean recovery is complete - children should continue outpatient follow-up.
Complications of SAM
| Complication | Notes |
|---|---|
| Hypoglycemia | Most common cause of early death; may be asymptomatic (lethargy only) |
| Hypothermia | Reduced metabolic rate, loss of insulating fat |
| Sepsis | Impaired immunity; may present without fever |
| Severe anemia | Multifactorial (iron, folate, protein, chronic infection) |
| Refeeding syndrome | Electrolyte shifts (hypophosphatemia, hypokalemia, hypomagnesemia) on rapid refeeding - reason for slow F-75 start |
| Heart failure | Risk with IV fluids or rapid fluid repletion |
| Infections | Measles, TB, malaria, pneumonia all worsen prognosis significantly |
| Diarrhea | SAM increases case fatality of diarrhea 9x |
| Developmental delay | Impaired brain myelination with early-onset SAM |
Community-Based Management (CMAM)
Uncomplicated SAM without medical complications can be managed at Outpatient Therapeutic Programs (OTP):
- RUTF given weekly at health facility
- Regular MUAC/weight monitoring
- Caregiver education on feeding and hygiene
- Treatment of simple infections (amoxicillin 7-day course)
- Has dramatically expanded SAM treatment coverage in resource-limited settings
Prevention (WHO/FAO Framework)
- Exclusive breastfeeding for 6 months, continued to 2 years
- Appropriate complementary feeding (introducing protein and energy-rich foods at 6 months)
- Immunization (especially measles, which precipitates kwashiorkor)
- Growth monitoring with regular weight charting
- Vitamin A supplementation (6-monthly in deficient populations)
- Zinc supplementation to reduce diarrhea severity/duration
- Safe water, sanitation, and hygiene
- Treatment of intestinal parasites
- Family planning and birth spacing
Park's Textbook of Preventive and Social Medicine
Key Points Summary
- SAM diagnosis: WHZ <-3 SD, MUAC <115 mm (field cut-off), or bilateral pitting edema
- Marasmus = calorie + protein deficiency; somatic wasting; no edema; albumin near-normal
- Kwashiorkor = predominantly protein deficiency; edema; fatty liver; hypoalbuminemia; skin and hair changes
- Management: WHO 10-step protocol in two phases (stabilization with F-75, then rehabilitation with F-100/RUTF)
- Critical early dangers: hypoglycemia, hypothermia, sepsis, refeeding syndrome, cardiac overload from IV fluids
- Do NOT give iron in Phase 1; do NOT give standard ORS (use ReSoMal); do NOT give IV fluids unless in shock
- Community-based RUTF programs have transformed SAM care globally
Sources:
- Robbins & Kumar Basic Pathology, Chapter 7
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 9
- Park's Textbook of Preventive and Social Medicine, Chapter on nutrition
Bronchopneumonia pediatrics
Reading File
Finding Sources
Finding Sources
Searching PubMed
pneumonia children management antibiotic
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
I have sufficient content from textbooks, plus the Fishman's and Robbins pathology sections. Let me now compile the comprehensive answer using all collected material plus my knowledge base for the pediatric-specific management aspects (WHO IMCI classification, age-specific pathogens, antibiotic choices).
Bronchopneumonia in Pediatrics
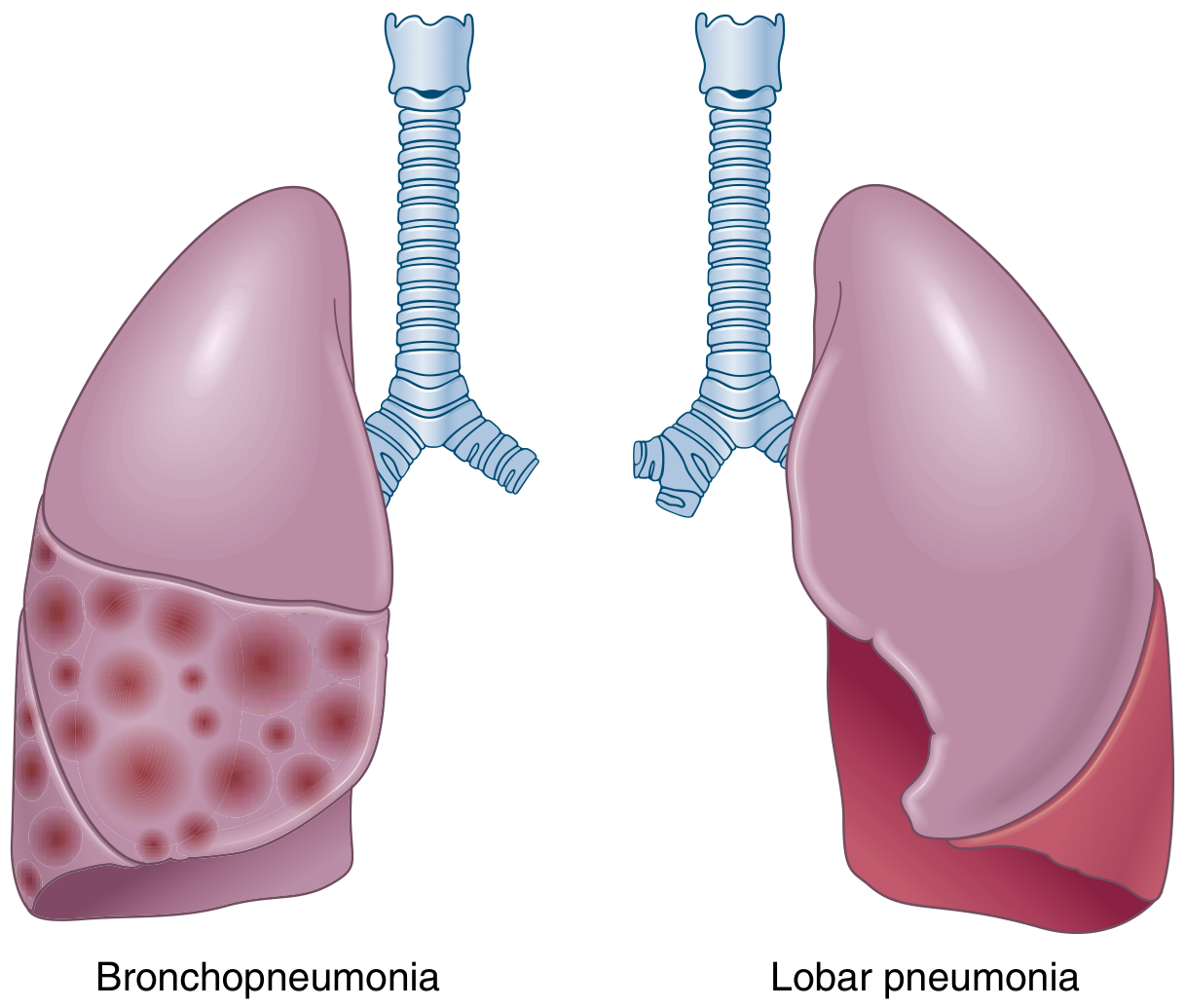
Definition
Bronchopneumonia (lobular pneumonia) is an acute suppurative inflammation of the lung characterized by patchy, multifocal consolidation originating in the bronchi and bronchioles and extending into the surrounding alveolar spaces. It is the most common form of pneumonia in infants and young children.
It differs from lobar pneumonia in that:
- Lobar pneumonia: homogeneous consolidation of an entire lobe or large portion
- Bronchopneumonia: patchy, segmental, bilateral, often basal consolidation
Robbins & Kumar Basic Pathology, p. 469
Anatomy of Distribution
Epidemiology and Importance
- One of the leading causes of death in children under 5 years globally
- Pneumonia accounts for approximately 14% of all deaths in children under 5
- Most deaths occur in low- and middle-income countries
- RSV is the most common cause in infants; S. pneumoniae predominates in older children
Etiology by Age Group
Age determines the most likely pathogen - a critical principle in pediatric practice:
| Age Group | Common Pathogens |
|---|---|
| Neonates (<1 month) | Group B Streptococcus (GBS), E. coli, Klebsiella, Listeria monocytogenes |
| 1-3 months | S. pneumoniae, H. influenzae, RSV, Chlamydia trachomatis (afebrile pneumonia), Bordetella pertussis |
| 4 months - 5 years | RSV, parainfluenza virus, human metapneumovirus, adenovirus, S. pneumoniae, H. influenzae, S. aureus, Mycoplasma pneumoniae (from ~3 years) |
| >5 years / school-age | S. pneumoniae, Mycoplasma pneumoniae (most common atypical; "walking pneumonia"), Chlamydophila pneumoniae, influenza |
Additional organisms of note:
- S. aureus: produces severe necrotizing bronchopneumonia; common post-influenza; associated with lung abscess and pneumatoceles
- H. influenzae (non-encapsulated): produces bronchopneumonic pattern
- Viral: RSV, metapneumovirus, parainfluenza, influenza, adenovirus - especially in children <2 years; damage to respiratory epithelium predisposes to secondary bacterial infection
- Viral and bacterial co-infection is common and worsens prognosis
Fishman's Pulmonary Diseases and Disorders; Robbins & Kumar Basic Pathology, Table 11.4
Pathogenesis
- Organisms reach the lower respiratory tract via inhalation (most common), aspiration of upper airway secretions, or hematogenous spread
- Infection begins in the bronchi and bronchioles (unlike lobar pneumonia which starts in alveoli)
- Epithelial ulceration of bronchiolar walls occurs
- Fibrinopurulent exudate (neutrophil-rich) fills the lumen and spreads into surrounding peribronchiolar alveolar spaces
- Impaired mucociliary clearance promotes spread; gravity pulls secretions to lower lobes (bilateral basal distribution)
- Viral infection particularly damages the respiratory epithelium → impairs mucociliary clearance → predisposes to secondary bacterial pneumonia
In children, predisposing factors include:
- Young age (immature immunity, smaller airways)
- Malnutrition (especially SAM - increases case fatality 9-fold)
- Lack of breastfeeding
- Indoor air pollution and smoke exposure
- Crowded living conditions
- Incomplete vaccination (measles, H. influenzae type b, S. pneumoniae vaccines)
- Prematurity and low birth weight
- Underlying conditions: congenital heart disease, CF, immune deficiency, sickle cell disease
Pathology (Morphology)
Gross:
- Focal areas of consolidation, slightly elevated, dry, granular, gray-red to yellow
- Often bilateral and basal (dependent areas due to secretion pooling)
- Multilobar involvement common
- Poorly demarcated margins (contrast with sharp lobar boundary in lobar pneumonia)
Histology:
- Neutrophil-rich exudate filling bronchi, bronchioles, and adjacent alveolar spaces
- Epithelial ulceration of bronchiolar walls
- Areas of consolidation: air replaced by suppurative exudate
- Unlike lobar pneumonia, no uniform staging (no red hepatization → gray hepatization)
Robbins & Kumar Basic Pathology, p. 469
Clinical Features
Symptoms
- Fever (high grade, often abrupt onset)
- Cough (may be productive in older children; often dry or tight in infants)
- Rapid breathing / tachypnea - the most sensitive and specific sign
- Difficulty breathing, respiratory distress
- Chest pain (pleuritic if pleuritis develops)
- Poor feeding, lethargy (especially in infants)
- Preceding upper respiratory tract infection (URTI) in many cases
Signs
- Tachypnea (see WHO respiratory rate thresholds below)
- Subcostal/intercostal recession (indrawing)
- Nasal flaring, grunting (especially in young infants)
- Reduced air entry on auscultation
- Crackles (crepitations) - fine inspiratory crackles at affected areas
- Bronchial breathing over consolidated segments
- Dullness to percussion over consolidated areas
- Fever, signs of toxicity
- Cyanosis (in severe disease)
- In infants: abdominal distension (swallowed air), poor feeding
WHO/IMCI Classification of Pneumonia in Children
The WHO Integrated Management of Childhood Illness (IMCI) classification is the globally used framework:
Respiratory Rate Thresholds for Tachypnea (WHO):
| Age | Fast Breathing Threshold |
|---|---|
| <2 months | ≥60 breaths/min |
| 2-11 months | ≥50 breaths/min |
| 1-5 years | ≥40 breaths/min |
| >5 years | ≥30 breaths/min |
Classification:
| Category | Criteria | Management |
|---|---|---|
| No pneumonia (cough/cold) | No tachypnea, no chest indrawing | Home care; symptomatic treatment |
| Pneumonia | Tachypnea present; no chest indrawing | Outpatient oral antibiotics; reassess in 2 days |
| Severe pneumonia | Tachypnea + chest indrawing | Hospital admission; oral/parenteral antibiotics; O2 if SpO2 <90% |
| Very severe pneumonia | Any danger sign: cyanosis, inability to drink, seizures, abnormal drowsiness/unconsciousness, severe respiratory distress, malnutrition | Urgent hospital/ICU admission; IV antibiotics; oxygen; supportive care |
General Danger Signs (any age):
- Cannot drink or breastfeed
- Vomits everything
- Convulsions
- Lethargic or unconscious
- Grunting (in young infants)
- Central cyanosis
Investigations
Chest X-Ray
- Bronchopneumonia: patchy, bilateral, multifocal opacities predominantly in lower zones; ill-defined margins; perihilar infiltrates
- Lobar consolidation: homogeneous opacity of an entire lobe
- Peribronchial thickening (especially with viral etiology)
- Hyperinflation suggests viral or bronchiolitic component
- Complications: pleural effusion, empyema, pneumatocele (especially S. aureus), abscess, pneumothorax
Note: CXR alone cannot reliably distinguish viral from bacterial pneumonia
Blood Tests
- CBC: leukocytosis with neutrophilia (bacterial); lymphocytosis (viral); leukopenia in severe sepsis
- CRP, procalcitonin: elevated in bacterial (procalcitonin >0.25-0.5 ng/mL favors bacterial)
- Blood culture: positive in only ~10-30% of bacterial pneumonia; should be obtained before antibiotics in hospitalized children
Microbiology
- Nasopharyngeal swab + multiplex PCR (most sensitive for viral etiology: RSV, influenza, metapneumovirus)
- Sputum (difficult to obtain in children <6 years)
- Urine pneumococcal antigen (useful in older children/adults)
- Pleural fluid culture and microscopy if effusion present
Pulse Oximetry
- Essential in all children with respiratory illness
- SpO2 <90% = significant hypoxemia; indicates need for oxygen therapy
Management
Outpatient Management (Non-severe Pneumonia)
Antibiotic choice (empirical):
| Age | First-line Antibiotic | Notes |
|---|---|---|
| 1-3 months | Refer to hospital | Always admit |
| 4 months - 5 years | Amoxicillin 40-90 mg/kg/day PO in 2-3 divided doses × 5 days | Covers S. pneumoniae; first-line per WHO |
| 5-15 years | Amoxicillin OR Azithromycin if atypical suspected | Mycoplasma common in school-age; azithromycin 10 mg/kg day 1, then 5 mg/kg days 2-5 |
| If penicillin allergic | Azithromycin or Clarithromycin |
- Give folic acid, ensure adequate hydration, nutrition
- Educate caregiver on danger signs: if no improvement in 48 hours OR develops danger signs → return immediately
- Reassess in 2 days
Inpatient Management (Severe/Very Severe Pneumonia)
1. Antibiotics (IV):
| Severity | Regimen |
|---|---|
| Severe pneumonia (no complications) | Ampicillin 50 mg/kg IV/IM q6h ± Gentamicin 7.5 mg/kg IV OD (WHO regimen) |
| Suspected S. aureus (pneumatocele, cavitation) | Add Cloxacillin (or Flucloxacillin) IV; or Vancomycin if MRSA suspected |
| Atypical pathogens suspected (>3 years) | Add Azithromycin IV/PO |
| Infants <3 months | Ampicillin + Gentamicin (covers GBS, gram-negatives) |
| Neonates | Ampicillin + Gentamicin (or Cefotaxime) |
| Oral step-down after 48-72 hrs of improvement | Amoxicillin PO; total duration 5-10 days |
2. Oxygen therapy:
- Indicated when SpO2 <90% (or <92% in children with underlying cardiac/lung disease)
- Deliver via nasal prongs, face mask, or headbox (infants)
- Target SpO2 ≥94%
- High-flow nasal cannula (HFNC) for moderate-severe hypoxemia
3. Supportive care:
- Antipyretics (paracetamol/acetaminophen for fever and comfort; avoid ibuprofen if dehydrated)
- IV fluids if cannot take orally (avoid overhydration - risk of SIADH in pneumonia)
- NG tube feeding if poor oral intake
- Chest physiotherapy: not routinely recommended in acute phase
- Bronchodilators: only if wheeze present (suggests bronchiolitic component or reactive airway disease)
- Mucolytics/antihistamines: not recommended
4. Monitoring:
- Respiratory rate, heart rate, SpO2, temperature - at least every 4 hours
- Fluid balance
- Response to antibiotics expected within 48-72 hours
Complications
| Complication | Details |
|---|---|
| Parapneumonic effusion / Empyema | Exudative effusion → infected pleural fluid; requires thoracocentesis; antibiotics (MRSA cover if empyema); consider chest drain |
| Lung abscess | Especially with S. aureus, Klebsiella, anaerobes (aspiration); cavitation on CXR/CT; prolonged antibiotics 4-6 weeks |
| Pneumatocele | Thin-walled air-filled cyst; characteristic of S. aureus pneumonia in children; usually resolves spontaneously |
| Pneumothorax | May occur with necrotizing pneumonia or tension in ventilated patients |
| Bacteremia / Septicemia | Secondary seeding: meningitis, endocarditis, septic arthritis, pericarditis |
| Respiratory failure | Requires CPAP or mechanical ventilation |
| SIADH | Inappropriate ADH secretion in severe pneumonia → hyponatremia; restrict fluids |
| Organization | Failure of exudate resorption → fibrosis; more common in S. aureus pneumonia |
| Empyema necessitans | Rare; pus tracks through chest wall |
Bronchopneumonia vs Lobar Pneumonia: Comparison
| Feature | Bronchopneumonia | Lobar Pneumonia |
|---|---|---|
| Distribution | Patchy, multifocal, bilateral, basal | Entire lobe, homogeneous |
| Origin | Bronchi/bronchioles | Alveoli |
| Pattern | Lobular | Lobar |
| X-ray | Patchy infiltrates, perihilar | Homogeneous lobar opacity |
| Common age | Infants, young children, elderly | Older children, adults |
| Common organisms | S. aureus, non-encapsulated H. influenzae, RSV, Mycoplasma, viral | S. pneumoniae (classic) |
| Exudate staging | No clear stages | 4 stages (congestion → red hepatization → gray hepatization → resolution) |
| Pleuritis | Less common | Common at periphery |
Specific Pneumonia Patterns in Children
Staphylococcal Pneumonia
- Severe, rapidly progressive bronchopneumonia
- Characteristic pneumatoceles (thin-walled air cysts) on CXR
- May develop empyema, pneumothorax, lung abscess
- Often follows influenza infection
- Requires anti-staphylococcal coverage (Cloxacillin IV; Vancomycin if MRSA)
Mycoplasma Pneumonia ("Walking Pneumonia")
- Most common atypical pathogen in school-age children
- Gradual onset; child often not very ill despite CXR changes
- Dry, hacking cough; myalgias; headache; low-grade fever
- CXR: diffuse bilateral patchy infiltrates, often worse than expected clinically
- Treatment: Azithromycin or Clarithromycin (macrolides); or Doxycycline in >8 years
Chlamydia trachomatis Pneumonia (Infants 1-3 months)
- Afebrile pneumonitis - characteristic feature
- Staccato cough, tachypnea, bilateral diffuse infiltrates
- Often with concurrent conjunctivitis
- Treatment: Erythromycin or Azithromycin (macrolide)
RSV Bronchiolitis-Pneumonia Overlap
- In infants <2 years; RSV is the dominant pathogen
- Wheeze, hyperinflation, bilateral peribronchial thickening
- Usually managed supportively (no proven benefit of antivirals in immunocompetent children)
Prevention
-
Vaccines - most effective intervention:
- Pneumococcal conjugate vaccine (PCV13/PCV15): major impact on S. pneumoniae pneumonia
- H. influenzae type b (Hib) vaccine: prevents Hib pneumonia/meningitis
- Measles vaccine: measles is a major predisposing cause of pneumonia in developing countries
- Influenza vaccine: annual, recommended from 6 months
- RSV prophylaxis: Nirsevimab (monoclonal antibody) for infants <12 months in first RSV season
-
Breastfeeding: exclusive breastfeeding for 6 months reduces pneumonia risk significantly
-
Nutrition: prevent malnutrition (especially vitamin A and zinc deficiencies)
-
Vitamin A supplementation: reduces pneumonia case fatality
-
Zinc supplementation: reduces incidence and duration of pneumonia
-
Reduce indoor air pollution: avoid cooking smoke, tobacco smoke exposure
-
Hand hygiene and sanitation
-
Zinc: reduces pneumonia incidence by ~41% in deficient populations
Key Points Summary
- Bronchopneumonia = patchy lobular consolidation starting in bronchioles; most common pneumonia pattern in infants and young children
- Etiology is age-dependent: viruses (RSV) dominate in infants; S. pneumoniae and Mycoplasma in older children
- Tachypnea is the most important clinical sign - use WHO age-specific thresholds
- WHO IMCI classifies severity based on respiratory rate + chest indrawing + danger signs
- Outpatient non-severe: oral amoxicillin; inpatient severe: IV ampicillin ± gentamicin
- S. aureus pneumonia: suspect if pneumatoceles, cavitation, or rapid deterioration → add anti-staphylococcal agent
- Atypical (Mycoplasma): school-age children, gradual onset, macrolide therapy
- Complications: empyema, abscess, pneumatocele, bacteremia, SIADH
- Prevention: PCV, Hib, measles vaccines + breastfeeding + nutrition are the pillars
Sources:
- Robbins & Kumar Basic Pathology, Chapter 11 (p. 469-471)
- Fishman's Pulmonary Diseases and Disorders, 2-Volume Set, Chapter 122
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- Goldman-Cecil Medicine, International Edition
- Harriet Lane Handbook, 23rd Edition (Johns Hopkins)
Nephrotic syndrome in pediatrics
Reading File
Finding Sources
Searching PubMed
nephrotic syndrome children treatment steroid
Reading File
Reading File
Reading Article
PMID: 40254362
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
I now have comprehensive content from multiple authoritative sources plus the KDIGO 2025 guideline. Let me compile the complete answer.
Nephrotic Syndrome in Pediatrics
Definition
Nephrotic syndrome (NS) is defined by the tetrad of:
| Feature | Threshold |
|---|---|
| Proteinuria (heavy) | >40 mg/m²/hr (or urine protein:creatinine ratio >2.0 mg/mg; or >3.5 g/24hr in adults) |
| Hypoalbuminemia | Serum albumin <2.5 g/dL (some sources <3.5 g/dL) |
| Generalized edema | Pitting edema, periorbital puffiness, ascites, scrotal/labial edema |
| Hyperlipidemia + lipiduria | Elevated total cholesterol, LDL; fatty casts in urine |
- In children, the most common presentation is steroid-sensitive nephrotic syndrome (SSNS), with minimal change disease (MCD) as the underlying lesion
- Hypertension is not typical of minimal change NS; its presence suggests a different underlying etiology
Campbell-Walsh Urology; National Kidney Foundation Primer on Kidney Diseases, 8e
Epidemiology
- Incidence: ~2-7 per 100,000 children per year
- Peak age: 2-6 years (preschool; median 2.5 years)
- Male:female ratio in young children: ~2:1 (equalizes in adolescence)
- Minimal change disease (MCD) accounts for:
- ~90% of cases in children <10 years
- ~70-80% of all pediatric NS overall
- FSGS is more prevalent in African-Americans, Hispanics, and Asians with NS
Classification
By Etiology
1. Congenital / Infantile NS (<3 months of age)
- Usually genetic (mutations in NPHS1 [nephrin], NPHS2 [podocin], WT1, LAMB2)
- Finnish type: most common; massive proteinuria from birth; placental enlargement
- Immunosuppression is not effective - managed conservatively then transplantation
- Infective causes: congenital syphilis, CMV, rubella, toxoplasmosis, HIV
2. Primary (Idiopathic) NS
- MCD, FSGS, mesangial proliferation, membranous nephropathy
- No identifiable systemic cause
3. Secondary NS
- Accounts for ~10% in children
- Causes: lupus nephritis (SLE), IgA nephropathy, Henoch-Schönlein purpura (IgA vasculitis), hepatitis B/C, malaria, HIV, amyloidosis, malignancy (Hodgkin lymphoma → MCD)
By Histological Lesion (Primary NS)
| Histology | Frequency in Children | Steroid Response |
|---|---|---|
| Minimal Change Disease (MCD) | ~70-90% | ~90% respond |
| Focal Segmental Glomerulosclerosis (FSGS) | <15% | <25% respond |
| Mesangial Proliferation | ~5-10% | Variable |
| Membranous Nephropathy (MN) | Rare in children | Variable |
By Steroid Response (Clinical Classification)
| Category | Definition |
|---|---|
| Steroid-sensitive NS (SSNS) | Complete remission (urine protein negative) within 8 weeks of standard steroid therapy |
| Steroid-resistant NS (SRNS) | Failure to achieve remission after 6-8 weeks of full-dose daily prednisone (15-20% of cases) |
| Frequently-relapsing NS (FRNS) | ≥2 relapses within 6 months of initial response, OR ≥4 relapses in any 12-month period |
| Steroid-dependent NS (SDNS) | Two consecutive relapses during steroid taper, OR requires continuous corticosteroids to maintain remission |
Campbell-Walsh Urology, p. 459; KDIGO 2025 Guideline
Pathophysiology
Why Proteinuria Occurs
- Normal glomerular filtration barrier consists of: fenestrated endothelium + glomerular basement membrane (GBM) + podocyte foot processes (filtration slits bridged by nephrin)
- In NS, loss of podocyte foot process architecture ("foot process effacement") disrupts the filtration barrier → massive protein leakage
- In MCD: T-cell-derived circulating permeability factor → podocyte injury → foot process effacement (visible only on electron microscopy - hence "minimal change" on light microscopy)
- In FSGS: progressive podocyte loss → segmental scarring of glomeruli
Pathophysiological Cascade
Podocyte injury
↓
Massive proteinuria (especially albumin)
↓
Hypoalbuminemia
↓
↓ Plasma oncotic pressure
↓
Fluid shifts to interstitium (underfill edema) + Primary renal Na⁺ retention (overfill)
↓
Generalized edema (periorbital, scrotal, ascites, pleural effusion)
↓
↑ Hepatic lipoprotein synthesis (response to ↓ oncotic pressure) + ↓ lipid clearance
↓
Hyperlipidemia (↑ cholesterol, LDL, VLDL) + Lipiduria (fatty casts)
- Underfill (especially MCD): low plasma volume → stimulated RAAS → secondary Na⁺ retention
- Overfill (other causes): primary renal Na⁺ retention → expanded plasma volume → suppressed RAAS
Brenner and Rector's The Kidney, p. 2277
Consequences of Urinary Protein Loss
| Lost Protein | Consequence |
|---|---|
| Albumin | Edema, hypoalbuminemia |
| Transferrin | Iron deficiency anemia |
| Vitamin D-binding protein | Vitamin D deficiency, hypocalcemia |
| Immunoglobulins | Hypogammaglobulinemia → susceptibility to infections |
| Antithrombin III, Protein C, Protein S, plasminogen | Hypercoagulable state → thrombosis |
| Thyroxine-binding globulin | Low T4 (usually euthyroid clinically) |
Clinical Features
Presentation
- Periorbital puffiness - often the earliest sign, noticed on waking; may be mistaken for allergy
- Dependent edema - legs, feet, sacral area
- Scrotal/labial edema (can be massive)
- Abdominal distension (ascites)
- Pleural effusion → dyspnea in severe cases
- Weight gain (fluid)
- Decreased urine output (concentrated, frothy urine)
- Child appears generally well initially (in MCD)
- No hypertension, no gross hematuria in typical MCD
On Examination
- Pitting edema (periorbital, peripheral, sacral)
- Abdominal fullness/fluid thrill (ascites)
- Normal blood pressure (in MCD)
- No rash (in idiopathic primary NS)
- Signs of underlying cause if secondary (butterfly rash in SLE, purpura in HSP)
Urine Appearance
- Frothy urine (protein)
- Lipid droplets ("oval fat bodies"), fatty casts ("Maltese crosses" under polarized light)
- Microscopic hematuria in ~20% (macroscopic hematuria is unusual - suggests different diagnosis)
Investigations
Urine
- Urine dipstick: 3+ or 4+ protein (screening; Albustix)
- Spot urine protein:creatinine ratio (PCR): >2.0 mg/mg (or >200 mg/mmol) = nephrotic range
- 24-hour urine protein: >40 mg/m²/hr in children
- Microscopy: fatty casts, oval fat bodies, lipid droplets
- Urine sodium: low (<20 mmol/L) in underfill state
Blood
- Serum albumin: <2.5 g/dL (often <1.5 g/dL in severe cases)
- Serum cholesterol: elevated (>200 mg/dL); LDL, VLDL elevated
- Serum electrolytes (may show hyponatremia from dilution)
- Serum creatinine and urea (usually normal in MCD; elevated in SRNS)
- Complement C3, C4: normal in MCD and primary NS; low C3 suggests postinfectious GN, MPGN; low C3+C4 suggests lupus
- ANA, anti-dsDNA, ANCA (to exclude SLE, vasculitis)
- Hepatitis B, C serology; HIV
- CBC: may show elevated hematocrit (hemoconcentration)
- Coagulation screen (hypercoagulable state)
Renal Biopsy
In children, a biopsy is NOT required before starting steroids if the presentation is typical:
- Age 1-12 years
- No hypertension
- No macroscopic hematuria
- Normal complement
- No systemic features (rash, arthritis)
Biopsy IS indicated if:
- Age <1 year (exclude congenital NS) or adolescent (higher chance of FSGS/MN)
- Macroscopic hematuria
- Persistent hypertension
- Low complement levels
- Extrarenal features (rash, arthritis)
- Steroid resistance (after 6-8 weeks of treatment)
- Atypical features
Histological Findings:
MCD on Light Microscopy: Normal glomeruli (hence "minimal change") - no proliferation, no deposits, no sclerosis
MCD on Electron Microscopy: Diffuse foot process effacement (podocyte "fusion") - the hallmark finding
FSGS on Light Microscopy: Segmental scarring/sclerosis in a focal number of glomeruli; podocyte hypertrophy at edges
National Kidney Foundation Primer on Kidney Diseases, 8e, p. 190
Management
Initial Treatment (First Presentation)
Corticosteroids are the cornerstone:
Prednisone/Prednisolone regimen (KDIGO 2021/2025):
- 60 mg/m²/day (max 60 mg/day) or 2 mg/kg/day (max 60 mg/day) as daily dose for 4-6 weeks
- Then switch to alternate-day dosing: 40 mg/m² alternate days for 2-3 months, with gradual taper over 5-6 months total
- Total initial course: at least 12 weeks (longer courses reduce relapse rates - KDIGO 2025 recommends 3-6 months)
Response criteria:
- Complete remission: urine dipstick trace/negative for 3 consecutive days (or urine PCR <0.2 mg/mg)
- ~90% of children with MCD achieve remission within 2-4 weeks of treatment
Relapse: urine protein 2+ or more for 3 consecutive days after prior remission
Relapse treatment: Same daily dose until remission, then taper
Steroid-Sparing Agents (for FRNS/SDNS)
When cumulative steroid toxicity becomes a concern:
| Agent | Indication | Dose / Notes |
|---|---|---|
| Levamisole | FRNS, SDNS (mild) | 2.5 mg/kg alternate days; cheap; reduces relapses; monitor for agranulocytosis |
| Cyclophosphamide | FRNS (better than SDNS) | 2-3 mg/kg/day PO for 8-12 weeks; total cumulative dose <168 mg/kg; gonadotoxicity with high cumulative dose; not for SDNS (poor response) |
| Mycophenolate mofetil (MMF) | FRNS, SDNS | 600 mg/m²/dose BD; well tolerated; GI side effects |
| Calcineurin inhibitors (CNIs) | FRNS, SDNS, SRNS | Tacrolimus (preferred): 0.1 mg/kg/day in 2 doses; Cyclosporine: 3-6 mg/kg/day; monitor trough levels and renal function; nephrotoxicity with long-term use; high relapse on discontinuation |
| Rituximab | SDNS, FRNS (resistant to above) | Anti-CD20 monoclonal antibody; 375 mg/m² IV × 1-4 doses; increasingly used; B-cell depletion; monitor for infection, PML |
KDIGO 2025 Guideline; Campbell-Walsh Urology, p. 459
Steroid-Resistant NS (SRNS)
- Defined: no remission after 6-8 weeks of full-dose daily prednisone
- Biopsy required (most will have FSGS)
- Check for genetic mutations (NPHS1, NPHS2, WT1, TRPC6, etc.) - ~30 genes identified
- Genetic SRNS: typically does not respond to any immunosuppression
- Treatment options:
- Calcineurin inhibitors (Tacrolimus or Cyclosporine) - first-line for non-genetic SRNS
- Add low-dose prednisone
- MMF as alternative or combination
- IV pulse methylprednisolone
- Rituximab in resistant cases
- ACE inhibitor / ARB - antiproteinuric effect regardless of response to immunosuppression
- Prognosis: up to 50% of SRNS children (especially FSGS) progress to end-stage kidney disease (ESKD) within 10 years
Congenital NS (<3 months)
- Immunosuppression is NOT effective - do not give steroids
- Conservative management:
- Sodium and fluid restriction
- Intermittent IV albumin + loop diuretics (furosemide) for severe edema
- Hypercaloric diet (nasogastric if needed)
- Thyroid hormone replacement (T4 lost in urine)
- ACE inhibitor/ARB to reduce proteinuria
- Prophylactic anticoagulation if severe (renal vein thrombosis risk)
- Bilateral nephrectomy → dialysis → renal transplantation (definitive treatment)
Membranous Nephropathy in Children
- Usually secondary (hepatitis B, SLE, PLA2R antibodies)
- ACE inhibitor/ARB + low-salt diet first
- Steroids + alkylating agents OR CNIs for progressive cases
- MMF and rituximab promising especially in PLA2R-antibody positive
Supportive Management
Edema
- Dietary sodium restriction (low-salt diet; <2 g Na/day)
- Fluid restriction only if severe hyponatremia
- Furosemide (loop diuretic): 1-2 mg/kg/dose PO/IV; for symptomatic edema
- Spironolactone (aldosterone antagonist): useful as adjunct (aldosterone elevated in underfill)
- IV 20% albumin infusion followed by furosemide: for severe symptomatic edema or refractory edema; not routinely recommended as it is transiently lost in urine
- A fractional sodium excretion <0.2% differentiates volume-contracted (caution with diuretics) from volume-expanded states
Brenner and Rector's The Kidney
Infection Prevention and Management
- Children with NS are immunocompromised (urinary loss of immunoglobulins + steroids)
- Spontaneous bacterial peritonitis (SBP): 2-6% incidence; most commonly S. pneumoniae and gram-negatives
- Suspect in any febrile child with NS and abdominal pain
- Diagnosis: paracentesis (PMN >250/mm³)
- Treatment: IV cefotaxime or ceftriaxone
- Pneumococcal vaccine: give before starting steroids if possible; revaccinate every 5 years
- Varicella (chickenpox): can be severe/fatal in children on high-dose steroids
- Varicella-exposed non-immune children on steroids → IV acyclovir or VZIg
- Stop steroids if varicella develops (if possible) and give IV acyclovir
- Avoid live vaccines while on high-dose steroids (>2 mg/kg/day for >2 weeks)
- Prophylactic penicillin V during active nephrotic syndrome (some centers)
Thromboembolism
- All nephrotic patients are hypercoagulable (loss of antithrombin III, protein C, protein S)
- Up to 20% experience thrombotic events (deep vein thrombosis, pulmonary embolism, renal vein thrombosis)
- Renal vein thrombosis: suspect with loin pain, hematuria, sudden worsening of renal function; especially with proteinuria >10 g/day and albumin <2 g/dL (membranous nephropathy carries highest risk)
- Prophylactic anticoagulation: considered in high-risk patients (albumin <2 g/dL, immobile, membranous nephropathy)
- Therapeutic anticoagulation (LMWH or warfarin) for established thrombosis
Hyperlipidemia
- Statin therapy if persistent nephrotic syndrome with prolonged hyperlipidemia
- Usually resolves with remission
Diet
- Low salt (sodium restriction essential)
- Normal protein intake (protein restriction is no longer recommended - it worsens hypoalbuminemia without benefit)
- Adequate calories to maintain growth
Steroid Side Effects to Monitor (Long-term)
- Growth retardation (alternate-day dosing minimizes this)
- Obesity, cushingoid features
- Hypertension (monitor regularly)
- Cataracts and glaucoma (ophthalmology review)
- Osteoporosis: calcium + vitamin D supplementation recommended
- Glycosuria/diabetes
- Behavioral changes
- Increased infection susceptibility
Complications Summary
| Complication | Notes |
|---|---|
| Infections | SBP, cellulitis, pneumococcal sepsis; leading cause of mortality |
| Thromboembolism | DVT, PE, renal vein thrombosis; ~20% of nephrotic patients |
| Acute kidney injury | From severe hypovolemia (underfill) or interstitial edema |
| Hypovolemic shock | Especially in MCD with aggressive diuresis |
| Anemia | Iron deficiency (transferrin loss), + anemia of CKD in SRNS |
| Vitamin D deficiency / rickets | Loss of vitamin D-binding protein |
| Hypothyroidism | Loss of TBG (usually subclinical) |
| ESKD | In SRNS, especially genetic FSGS; ~50% at 10 years |
| Growth failure | From disease + chronic steroid use |
| Steroid toxicity | Cataracts, hypertension, obesity, osteoporosis, avascular necrosis |
Prognosis
| Group | Prognosis |
|---|---|
| SSNS/MCD | Excellent; ~85-90% achieve long-lasting remission; ~30-40% never relapse again after first episode |
| FRNS/SDNS | Good long-term renal function but chronic relapsing course; risk of steroid toxicity |
| SRNS (non-genetic FSGS) | ~50% develop ESKD within 5-10 years |
| Genetic SRNS | Poor; ESKD typically in childhood; transplantation required (recurrence risk post-transplant in non-genetic FSGS) |
| Congenital NS | Death or ESKD without transplantation |
Key Points Summary
- Classic pediatric NS: 2-6 year old boy with periorbital puffiness and edema, frothy urine, normal BP, normal complement
- MCD accounts for ~90% of NS in children 1-10 years; diagnosis/treatment without biopsy is standard in typical presentation
- Steroid (prednisolone) 60 mg/m²/day × 4-6 weeks then alternate-day taper = first-line treatment
- ~90% of MCD responds; ~50% will have at least one relapse
- Biopsy is needed before treating SRNS (most will have FSGS)
- SRNS → genetic testing (30 known genes) + calcineurin inhibitors
- Hypercoagulability and infections (SBP, pneumococcal sepsis) are the most dangerous complications
- KDIGO 2025 guideline now emphasizes: longer initial steroid courses (3-6 months), updated algorithm for biopsy timing, and rituximab as effective steroid-sparing agent
Recent Evidence:
- KDIGO 2025 Clinical Practice Guideline (Floege et al., Kidney Int 2025, PMID 40254362) - now includes updated treatment algorithms for steroid-sensitive, frequently-relapsing, steroid-dependent, and steroid-resistant NS, incorporating rituximab as a primary steroid-sparing agent
- Cochrane Review on corticosteroid therapy for NS in children (Hahn et al., Cochrane Database Syst Rev 2024, PMID 39171624) - confirms prolonged initial therapy reduces relapse rates
- Cochrane Review on steroid-resistant NS in children (Liu et al., Cochrane Database Syst Rev 2025, PMID 40337980) - updated evidence for CNIs, MMF, rituximab
Sources:
- Campbell-Walsh Urology, 3-Volume Set (Chapter on Nephrotic Syndrome in children)
- Brenner and Rector's The Kidney, 2-Volume Set
- National Kidney Foundation Primer on Kidney Diseases, 8e
- KDIGO 2025 Clinical Practice Guideline for the Management of Nephrotic Syndrome in Children
Normal new born pediatrics
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I have excellent content and images. Now let me compile the comprehensive answer on the normal newborn.
The Normal Newborn in Pediatrics
Definition: Periods and Terms
- Neonatal period: first 28 days of life (0-28 days)
- Newborn/neonate: birth to 28 days
- Term infant: 37-42 completed weeks of gestation
- Preterm: <37 weeks; Post-term: >42 weeks
- Normal birth weight: 2,500-4,000 g
1. Anthropometry at Birth (Normal Term Infant)
| Parameter | Normal Range | Notes |
|---|---|---|
| Birth weight | 2,500 - 4,000 g (average ~3,200-3,400 g) | <2,500 g = LBW; <1,500 g = VLBW; <1,000 g = ELBW |
| Length | 48 - 52 cm (average ~50 cm) | |
| Head circumference | 33 - 37 cm (average ~34-35 cm) | OFC (occipitofrontal circumference) |
| Chest circumference | 30 - 34 cm (average ~32-33 cm) | Head circumference > chest by 1-2 cm at birth |
Weight Classification by Gestational Age
- AGA (Appropriate for gestational age): weight between 10th and 90th percentile
- SGA (Small for gestational age): weight <10th percentile - at risk for hypoglycemia, temperature instability
- LGA (Large for gestational age): weight >90th percentile - often born to mothers with uncontrolled diabetes; at risk for hypoglycemia, shoulder dystocia, birth trauma
Textbook of Family Medicine 9e, p. 527
2. Vital Signs (Normal Term Newborn)
| Parameter | Normal Range |
|---|---|
| Heart rate | 120-160 beats/min (may drop to 90-100 during sleep; may reach 180 when crying) |
| Respiratory rate | 40-60 breaths/min |
| Temperature (axillary) | 36.5-37.5°C |
| Blood pressure | Systolic 60-90 mmHg; Diastolic 20-60 mmHg |
| SpO2 (preductal, right wrist) | Reaches ~90% by 10 minutes of life (initially 60% at 1 min is normal) |
- Tachypnea = respiratory rate >60 breaths/min
- Normal newborns may have periodic breathing (irregular respiratory rhythm with brief pauses up to 10 seconds) - normal; distinguish from true apnea (>20 sec pause)
3. Apgar Score
Assessed at 1 minute (need for resuscitation) and 5 minutes (efficacy of resuscitation). If score <7 at 5 minutes, repeat every 5 minutes up to 20 minutes.
| Sign | 0 | 1 | 2 |
|---|---|---|---|
| Heart rate | Absent | <100 beats/min | ≥100 beats/min |
| Respirations | Absent | Slow, irregular | Strong, crying |
| Muscle tone | Limp | Some flexion | Good flexion, active motion |
| Reflex irritability (to tactile stimulation) | No response | Grimace | Cough, sneeze, cry |
| Color | Blue/pale | Pink body, blue extremities | Completely pink |
Score interpretation:
- 7-10: Normal
- 4-6: Moderate depression; stimulation and/or supplemental oxygen
- 0-3: Severe depression; immediate resuscitation required
A low Apgar score at 1 minute is not predictive of long-term neurologic outcome. A score of 0-3 at 5 minutes correlates with increased risk of neonatal death. Resuscitation must NOT be interrupted to calculate the Apgar score.
The mnemonic Appearance, Pulse, Grimace, Activity, Respiration helps recall the five components.
Textbook of Family Medicine 9e; Rosen's Emergency Medicine
4. Gestational Age Assessment: Ballard Score
The New Ballard Score assesses both neuromuscular and physical maturity criteria (range -10 to +50; score correlates with gestational age in weeks).
Neuromuscular Maturity Criteria:
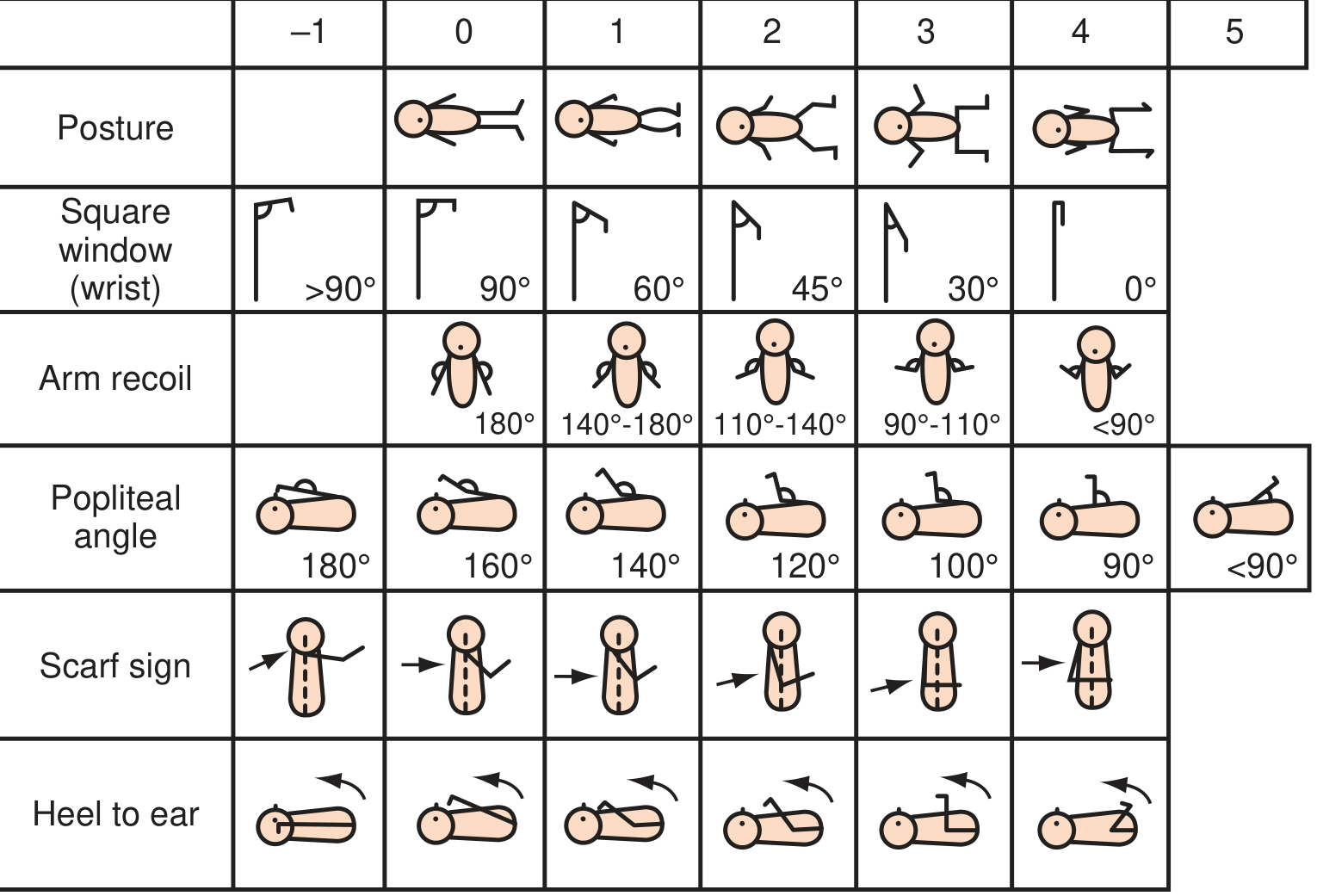
| Criterion | Preterm (immature) | Term (mature) |
|---|---|---|
| Posture | Floppy, extended | Strong flexion of all limbs |
| Square window (wrist) | >90° | 0° (wrist fully flexes to forearm) |
| Arm recoil | 180° (stays extended) | <90° (springs back) |
| Popliteal angle | 180° (fully extendable) | <90° |
| Scarf sign | Elbow crosses midline | Elbow does not reach midline |
| Heel to ear | Heel easily reaches ear | Resistance; cannot reach ear |
Physical Maturity Criteria:
| Criterion | Preterm | Term |
|---|---|---|
| Skin | Sticky, friable, transparent | Thick, cracking, parchment; little superficial peeling |
| Lanugo | None (very preterm), then abundant | Mostly on shoulders/bald areas |
| Plantar surface | Heel-toe <40 mm | Creases over entire sole |
| Breast | Imperceptible | Full areola, 5-10 mm bud |
| Eyes/ears | Eyelids fused; ear flat, stays folded | Eyes open; ear thick, instant recoil |
| Genitalia (male) | Scrotum flat, smooth | Testes descended; deep rugae |
| Genitalia (female) | Clitoris prominent; labia flat | Majora covers minora and clitoris |
Textbook of Family Medicine 9e, p. 527-528
5. Physical Examination of the Normal Newborn
General
- Well-flexed posture; symmetric movements
- Vigorous cry; responsive to stimuli
- Skin may show acrocyanosis (blue hands and feet) in first few hours - normal; central cyanosis is abnormal
- Vernix caseosa: white, greasy material covering the skin at birth (more in preterm)
Head
- Molding: overlapping of skull bones from passage through birth canal; resolves in 48-72 hours
- Caput succedaneum: diffuse, edematous swelling of the scalp; crosses suture lines; present at birth; resolves in 1-3 days
- Cephalhematoma: subperiosteal hemorrhage; does NOT cross suture lines; appears after birth; may take weeks to resolve; associated with jaundice
- Anterior fontanelle: diamond-shaped; normally soft and flat; 1-4 cm; closes at 12-18 months
- Posterior fontanelle: triangular; closes by 6-8 weeks
- Skull sutures: sagittal, coronal, lambdoid, metopic - should be palpable; note any premature fusion (craniosynostosis)
Eyes
- Intermittent crossing of the eyes is normal in first few months (immature ocular control)
- Red reflex must be present bilaterally - absence suggests cataract, retinoblastoma, retinal detachment → urgent ophthalmology referral
- Subconjunctival hemorrhages: from birth trauma; resolve spontaneously
- Pupils equal and reactive to light
- Doll's eye reflex (oculocephalic reflex): head turns → eyes move opposite direction; present in newborns
Ears, Nose, Throat
- Ears: check position (low-set ears suggest chromosomal abnormality); pinna recoils instantly in term infant
- Check patency of nares (choanal atresia: obstruction → respiratory distress; test by passing small catheter)
- Epstein pearls: small white retention cysts on hard palate - normal; resolve in weeks
- Bohn nodules: similar cysts on gum margins - normal
- Palate must be palpated to exclude submucous cleft (not visible to inspection alone)
- Check for ankyloglossia (tongue tie): short frenulum; may affect breastfeeding
Chest and Lungs
- Gynecomastia in both male and female infants (maternal estrogen): normal; may express "witch's milk" (white milky discharge) - normal; resolves within weeks
- Clavicles: palpate for fracture (crepitus, asymmetric Moro, tenderness) - especially after shoulder dystocia
- Normal breath sounds bilateral and equal
- Tachypnea (RR >60) requires evaluation; grunting, nasal flaring, retractions indicate respiratory distress
Cardiovascular
- Heart rate 120-160 bpm
- Femoral pulses: must be palpated bilaterally; absent/weak femoral pulses suggest coarctation of the aorta
- Systolic murmur may be present in first 24-48 hours due to closing ductus arteriosus - usually benign; persistent murmur needs evaluation
- Patent ductus arteriosus (PDA): continuous "machinery" murmur; more common in preterm
- Four-limb blood pressure and pulse oximetry (right hand + foot) to screen for critical congenital heart disease
Abdomen
- Appears slightly full/rounded; soft
- Liver normally palpable 1-2 cm below costal margin (normal)
- Spleen tip may be barely palpable
- Kidneys may be palpable by deep palpation in first day
- Umbilical cord: 3 vessels (2 arteries + 1 vein); single umbilical artery (2-vessel cord) is associated with chromosomal abnormalities and renal/cardiac anomalies - evaluate further
- Cord separates in 7-14 days; keep dry; no routine alcohol recommended (delays separation)
Genitalia
Male:
- Testes: both fully descended into scrotum in term male
- Check for undescended testis (cryptorchidism): if unilateral and not descended by 6 months, refer for orchidopexy
- Hypospadias: urethral opening on ventral surface of penis (glanular most common); if present, do NOT circumcise (foreskin may be used in repair)
- Assess for hydrocele: transilluminates; usually resolves by 1 year
- Phimosis: physiological and normal in newborn (non-retractile foreskin)
Female:
- Labia majora covers minora in term females
- Vaginal discharge (white mucoid) and/or blood-tinged discharge: normal in first 2 weeks; due to maternal estrogen withdrawal ("pseudo-menstruation")
- Labial adhesions: thin film of tissue between labia minora; benign, resolves spontaneously
- Clitoromegaly suggests congenital adrenal hyperplasia (CAH) in an XX infant
Spine and Back
- Inspect full spine for neural tube defects: hairy patches, sacral dimples, soft tissue masses, hemangioma, skin discoloration
- Sacral dimples: common; those with intact base within 2.5 cm of anus are benign; deep, large, or non-visible-base dimples require spinal ultrasound
Hips - DDH Screening
Developmental dysplasia of the hip (DDH):
- Risk factors: female sex, breech presentation, family history, oligohydramnios, first-born
- All newborns screened by:
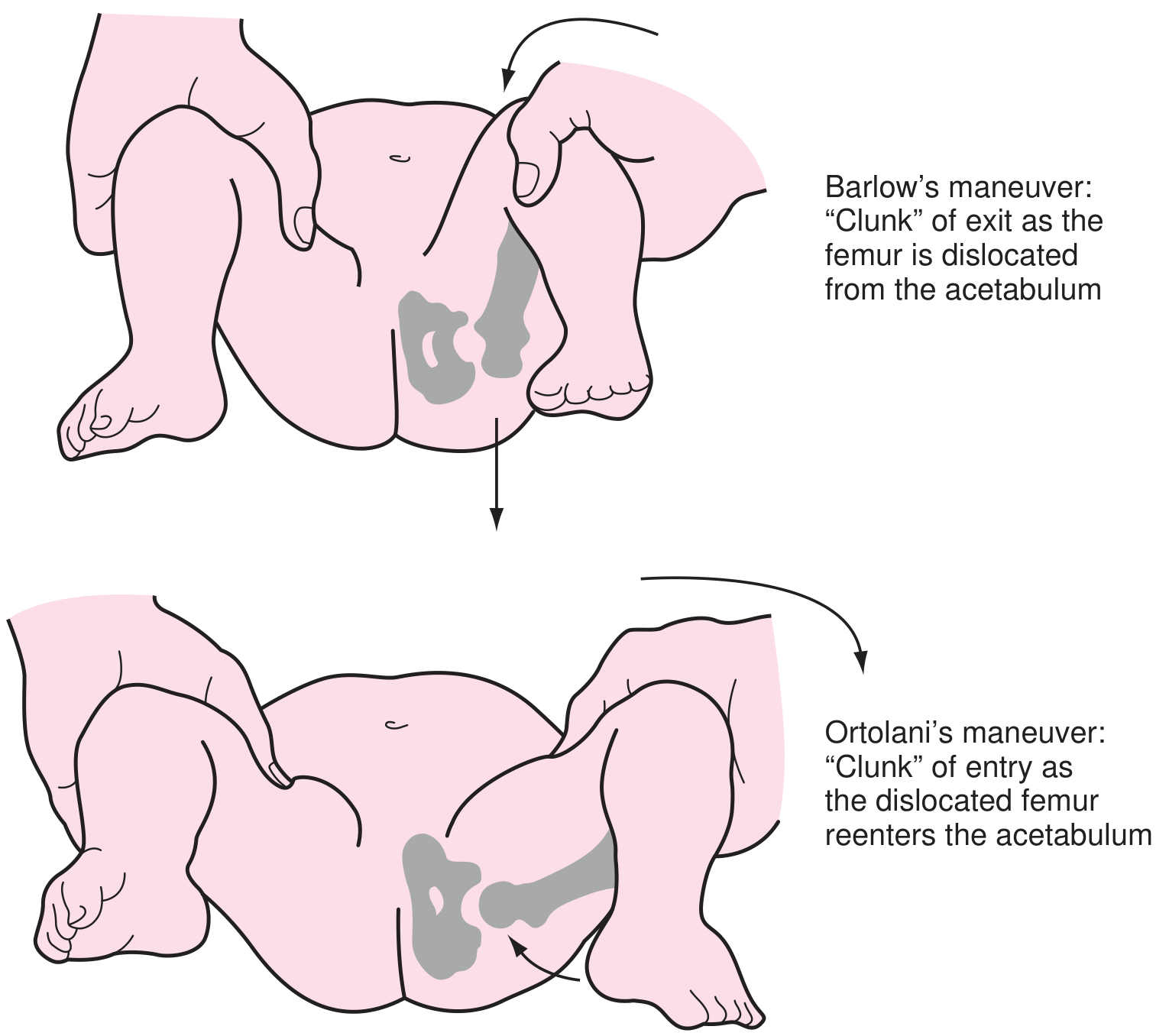
- Ortolani maneuver: hip ABduction + pressure on greater trochanter → "clunk" as dislocated femoral head relocates = positive (reduces a dislocated hip)
- Barlow maneuver: hip ADduction + posterior pressure → "clunk" as femoral head dislocates = positive (dislocates an unstable hip)
Management:
- Positive exam → refer orthopedics
- Equivocal → re-examine at 2-week visit
- Breech (normal exam) or female with family history → hip ultrasound at 6 weeks
Skin - Normal Findings and Transient Conditions
| Finding | Description | Significance |
|---|---|---|
| Vernix caseosa | White, greasy coating | Protective; normal |
| Milia | Tiny white papules on nose/cheeks (blocked sebaceous glands) | Normal; resolve in weeks |
| Erythema toxicum neonatorum | Blotchy erythematous rash with central pustule; "flea-bite" appearance; anywhere except palms/soles | Most common rash; benign; resolves in 1-2 weeks; smear shows eosinophils |
| Mongolian spots | Blue-gray patches over sacrum/buttocks | Normal in dark-skinned infants; may be mistaken for bruising; disappear by school age |
| Acrocyanosis | Blue discoloration of hands and feet | Normal in first hours; central cyanosis is abnormal |
| Harlequin color change | One half of body turns red, other pale; sharp midline demarcation | Benign vasomotor phenomenon; lasts minutes |
| Salmon patches / "stork bites" | Pink-red flat capillary hemangiomas on nape, eyelids, forehead | Very common; most fade by 2 years; those on nape may persist |
| Port wine stain (nevus flammeus) | Deep red-purple; does NOT fade; if over trigeminal distribution → suspect Sturge-Weber syndrome | Permanent; consider neurological evaluation |
| Cutis marmorata | Mottled, reticulated bluish-red discoloration on cooling | Normal vasomotor response; persists in hypothyroidism/trisomy 21 |
| Lanugo | Fine, downy hair; abundant in preterm | Normal |
6. Neurological Examination
Tone
- Term infant: strong flexion of all four extremities at rest
- Hypotonia ("floppy infant"): lies in frog-legged position; suggests Down syndrome, hypothyroidism, birth asphyxia, spinal muscular atrophy
- Hypertonia: spasticity or rigid movements; suggests HIE, drug withdrawal, meningitis
Primitive (Neonatal) Reflexes
| Reflex | How to Elicit | Response | Age Disappears |
|---|---|---|---|
| Moro (startle) reflex | Sudden head drop or loud noise | Symmetric abduction and extension of arms, then adduction ("embrace"); crying | 4-6 months |
| Rooting reflex | Stroke cheek/corner of mouth | Turns head toward stimulus; opens mouth | 3-4 months (while awake) |
| Sucking reflex | Place finger/nipple in mouth | Rhythmic sucking | 4 months (voluntary takes over) |
| Palmar grasp | Place finger in palm | Finger flexion around examiner's finger | 4-6 months |
| Plantar grasp | Pressure on ball of foot | Toe flexion | 9-10 months |
| Stepping (walking) reflex | Hold upright; foot touches surface | Alternating stepping movements | 2 months |
| Tonic neck reflex (TNR) / "Fencing" | Turn head to one side | Ipsilateral arm/leg extend; contralateral flex | 4-6 months |
| Babinski reflex | Stroke lateral sole upward | Extension (dorsiflexion) of big toe + fan of other toes | Becomes plantar flexion by 12-18 months (abnormal if persists) |
| Galant (trunk incurvation) reflex | Stroke paravertebral skin | Lateral curvature toward stimulus | 2 months |
| Parachute reflex | Held horizontally, lowered toward surface | Arms extend outward to "break fall" | Appears at 8-9 months; persists lifelong |
Key points:
- Asymmetric or absent primitive reflexes suggest neurological injury (birth trauma, nerve palsy, CNS lesion)
- Asymmetric Moro: suspect brachial plexus injury (Erb's palsy - C5-C6) or fractured clavicle
- Reflexes disappear as cortical control matures; persistence beyond expected age suggests CNS pathology
Textbook of Family Medicine 9e, Table 21-5
7. Physiological Adaptations at Birth
Cardiovascular Transition
-
In utero: lungs are fluid-filled; gas exchange via placenta; pulmonary vascular resistance (PVR) is high; blood bypasses lungs via:
- Foramen ovale (RA → LA)
- Ductus arteriosus (PA → aorta)
- Ductus venosus (umbilical vein → IVC)
-
At birth:
- First breath → lungs expand → PVR falls dramatically
- Increased pulmonary blood flow → increased LA pressure → foramen ovale closes (functional closure)
- Rise in PaO2 → ductus arteriosus constricts → closes functionally in 12-24 hours; permanent closure in 2-3 weeks
- Umbilical arteries spasm → ductus venosus closes
-
Preterm infants have higher risk of PDA persisting (lower oxygen sensitivity)
Respiratory Transition
- Fetal lung fluid is resorbed at birth (via squeezing in birth canal, lymphatics, circulatory absorption)
- Surfactant (Type II pneumocytes) essential for alveolar stability; production matures at ~34-36 weeks
- Surfactant deficiency → Respiratory Distress Syndrome (RDS) in preterm
Temperature Regulation
- Newborns are obligate heat losers (large surface area-to-volume ratio; poor insulation; limited brown fat mobilization in very preterm)
- Heat loss mechanisms: radiation (most important), conduction, convection, evaporation
- Term newborn can generate heat via non-shivering thermogenesis (brown adipose tissue/BAT metabolism)
- Target temperature: 36.5-37.5°C axillary; neutral thermal environment for preterm = incubator
Glucose Homeostasis
- Fetal glucose entirely from mother; at birth → glucose supply abruptly cut
- Newborns must rapidly activate gluconeogenesis and glycogenolysis
- Normal glucose threshold: >2.6 mmol/L (>47 mg/dL) after 4 hours of life
- Early feeding is the cornerstone of neonatal hypoglycemia prevention
8. Physiological Events in the First Week (Normal Variants)
| Event | Time Course | Notes |
|---|---|---|
| Physiological weight loss | First 3-5 days: up to 10% of birth weight | From fluid loss; regain by 7-10 days |
| Physiological jaundice | Days 2-5 (term); days 3-7 (preterm) | Due to high RBC turnover + immature liver conjugation; peaks by day 3-4 in term; if appears <24 hours → pathological |
| Meconium (first stool) | First 24-48 hours | Dark green/black; sterile; failure to pass in 48 hrs → exclude Hirschsprung's, cystic fibrosis |
| Transitional stools | Days 3-5 | Green-yellow (as milk feeds establish) |
| First urine | Within 24 hours | May have pink-orange "brick dust" staining (urate crystals) - normal in first few days |
| Umbilical cord separation | Days 7-14 | Keep dry; "wet" cord care not recommended |
| Physiological breast enlargement | Days 2-4 | Both sexes; maternal estrogen; may express "witch's milk"; resolves weeks |
| Vaginal discharge/pseudomenstruation | Days 1-7 | Normal; maternal estrogen withdrawal |
9. Routine Newborn Care
Immediate After Birth
- Dry and warm the infant immediately (prevents hypothermia)
- Clear airway if needed (bulb syringe if secretions)
- Assess Apgar score at 1 and 5 minutes
- Delayed cord clamping: wait at least 30-60 seconds before clamping (improves iron stores, reduces IVH in preterm)
- Skin-to-skin contact (kangaroo care): promotes bonding, thermoregulation, breastfeeding
- Initiate breastfeeding within 1 hour of birth
Routine Prophylaxis (within first hours)
| Intervention | Purpose | Details |
|---|---|---|
| Vitamin K (IM) | Prevents hemorrhagic disease of the newborn (HDN/VKDB) | 0.5-1.0 mg IM; all newborns are born vitamin K deficient; IM preferred over oral (more effective for late VKDB) |
| Erythromycin 0.5% ophthalmic ointment | Prevents gonococcal ophthalmia neonatorum | Apply both eyes within first 24 hours; 28% of infants born to infected mothers develop it without prophylaxis |
| Hepatitis B vaccine | Prevents perinatal HBV transmission | First dose within 24 hours of birth (immediately if mother HBsAg+); if mother HBsAg+: also give HBIg within 12 hours |
Newborn Screening (varies by country)
- Metabolic screen (heel-prick/Guthrie test): at 48-72 hours of age (after feeding established)
- PKU (phenylketonuria), congenital hypothyroidism, galactosemia, CAH, maple syrup urine disease, homocystinuria, cystic fibrosis (IRT), sickle cell disease, biotinidase deficiency, fatty acid oxidation disorders, amino acid disorders
- Hearing screen (OAE or AABR): before discharge
- Pulse oximetry (CCHD screen): right hand and foot SpO2 after 24 hours
- Bilirubin (transcutaneous or serum): for jaundice assessment
Feeding
- Breastfeeding recommended exclusively for first 6 months (WHO)
- Colostrum (first 3-5 days): rich in IgA, leukocytes, proteins; immunological protection
- Normal feeding frequency: 8-12 feeds per day (on demand, approximately every 2-3 hours)
- Adequacy: 6+ wet diapers/day and regular stools after milk comes in (day 3-5)
10. Red Flags in the Newborn
Any of the following require urgent evaluation:
| Red Flag | Possible Cause |
|---|---|
| Jaundice <24 hours of age | Hemolytic disease (Rh/ABO incompatibility) |
| Persistent central cyanosis | Cyanotic congenital heart disease, severe RDS |
| Respiratory rate >60/min persisting | RDS, TTN, pneumonia, congenital heart disease |
| Absent or asymmetric femoral pulses | Coarctation of the aorta |
| Absent red reflex | Cataract, retinoblastoma, glaucoma |
| Failure to pass meconium in 48 hours | Hirschsprung's disease, CF, imperforate anus |
| Failure to pass urine in 24 hours | Posterior urethral valves, bilateral renal agenesis |
| Hypotonia | Down syndrome, HIE, hypothyroidism, SMA |
| Ambiguous genitalia | CAH, intersex conditions |
| Absent Moro/asymmetric Moro | Birth asphyxia, brachial plexus injury, clavicle fracture |
| Single umbilical artery | Associated renal/cardiac anomalies |
| Sacral dimple with non-intact base | Neural tube defect/spinal abnormality |
Key Points Summary
- Normal term newborn: weight 2,500-4,000 g, length 48-52 cm, OFC 33-37 cm
- Apgar score at 1 and 5 min; 7-10 = normal; do NOT delay resuscitation for score
- Gestational age assessed by New Ballard score (neuromuscular + physical criteria)
- Physiological weight loss up to 10% expected; regain by 7-10 days
- Physiological jaundice starts day 2-5; jaundice <24 hours = always pathological
- Routine prophylaxis: Vitamin K (IM), erythromycin eye drops, Hepatitis B vaccine
- Primitive reflexes all present at birth; disappear with cortical maturation
- DDH: screen with Barlow (dislocates) and Ortolani (reduces); "clunk" = positive
- Key red flags: early jaundice, central cyanosis, absent femoral pulses, absent red reflex, failure to pass meconium/urine
Sources:
- Textbook of Family Medicine, 9th Edition, Chapter 21 (The Normal Newborn)
- Rosen's Emergency Medicine - Concepts and Clinical Practice
- Tintinalli's Emergency Medicine: A Comprehensive Study Guide
This is a shared conversation. Sign in to Orris to start your own chat.