I have my university exam of 1 st year of subject biochemistry These are the important question give me answer of this "Phenylketonuria (PKU)" and remeber it's a university examination The answer should be easy to memorize and follow the pattern of answer writing as in University paper
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
I have all the information needed. Now let me compile a comprehensive, exam-ready answer:
Phenylketonuria (PKU)
Definition
Phenylketonuria (PKU) is an autosomal recessive inborn error of amino acid metabolism characterized by a deficiency of the enzyme phenylalanine hydroxylase (PAH), resulting in the inability to convert phenylalanine to tyrosine, leading to accumulation of phenylalanine and its toxic metabolites in the body.
Incidence / Epidemiology
- Occurs in approximately 1 in 10,000 live-born infants
- More common in persons of Scandinavian/Northern European descent
- Less common in African American and Jewish populations
- Equally affects both sexes (autosomal recessive)
Biochemical Basis / Pathogenesis
Normal Pathway:
Phenylalanine → (PAH + BH4) → Tyrosine → Melanin / other products
In a healthy individual, the enzyme Phenylalanine Hydroxylase (PAH), using the cofactor Tetrahydrobiopterin (BH4), converts phenylalanine to tyrosine. The cofactor BH4 is regenerated from dihydrobiopterin (BH2) by the enzyme Dihydropteridine Reductase (DHPR).
In PKU:
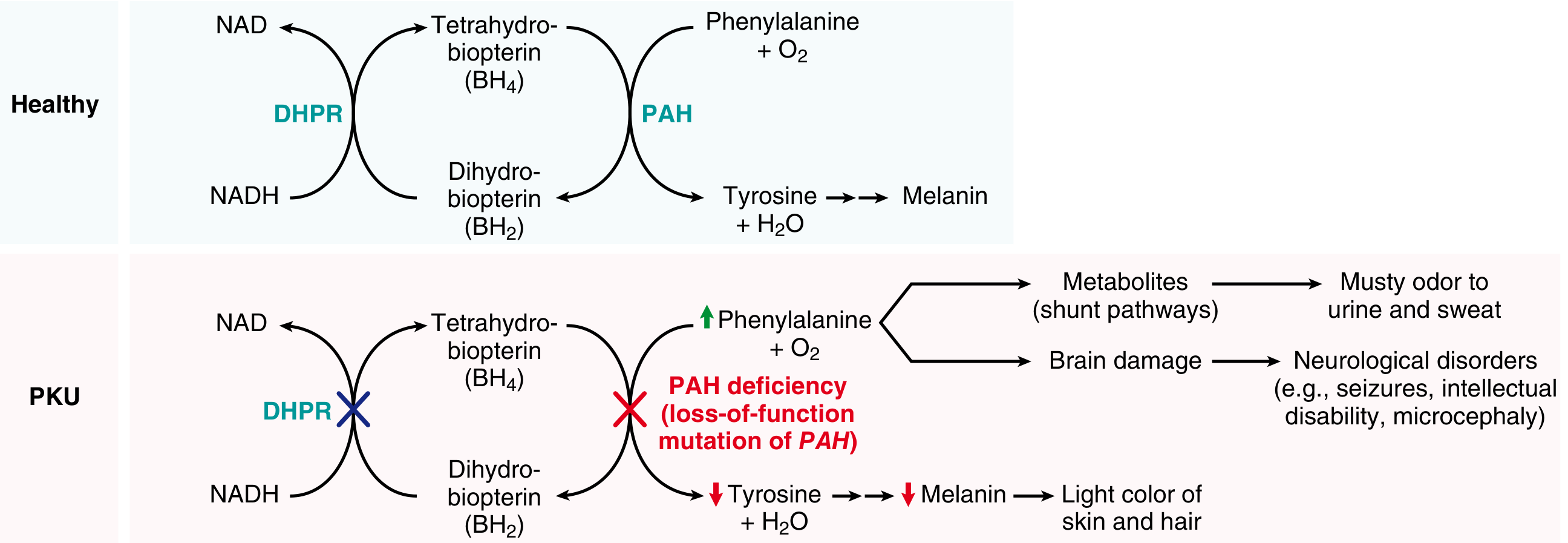
When PAH is deficient:
- Phenylalanine accumulates in blood (hyperphenylalaninemia)
- It is shunted into minor pathways producing abnormal metabolites:
- Phenylpyruvate (phenylketone - excreted in urine - gives the disease its name)
- Phenylacetate (gives musty/mousy odor to urine and sweat)
- Phenyllactate
- Tyrosine is deficient - since phenylalanine cannot be converted to tyrosine
- Tyrosine is a precursor for melanin - hence reduced pigmentation
These excess metabolites and phenylalanine itself cause brain damage by interfering with neurotransmitter synthesis and transport across the blood-brain barrier.
(Source: Robbins, Cotran & Kumar Pathologic Basis of Disease)
Types of PKU
| Type | Cause | Frequency |
|---|---|---|
| Classic PKU | Deficiency of PAH | ~98% of cases |
| Variant (Non-classic) PKU | Deficiency of DHPR OR inability to synthesize BH4 | ~2% of cases |
| Benign Hyperphenylalaninemia | Partial PAH deficiency - modest Phe elevation, no neurological damage | Milder form |
Clinical Features
In Untreated Infants / Children:
| Feature | Mechanism |
|---|---|
| Intellectual disability (severe) | Toxic effect of phenylalanine/metabolites on developing brain |
| Seizures | Neurological damage |
| Fair skin and hair (hypopigmentation) | Tyrosine deficiency → decreased melanin |
| Blue eyes | Reduced melanin |
| Musty/mousy odor | Phenylacetate in urine and sweat |
| Eczema | Skin manifestation |
| Microcephaly | Impaired brain development |
| Hyperreflexia, motor deficits | Neurological involvement |
"Fewer than 4% of untreated phenylketonuric children have IQs greater than 50 or 60; about one-third never walk and two-thirds cannot talk." - Robbins, Cotran & Kumar
Genetics
- Autosomal recessive inheritance
- Gene: PAH gene (chromosome 12q23)
- Almost 1000 mutant alleles of the PAH gene have been identified
- Mutations in both alleles are required (homozygous or compound heterozygous) to develop the disease
- Heterozygous carriers - phenotypically normal
Diagnosis
Neonatal Screening (Most important):
- Guthrie Test (Bacterial Inhibition Assay) - blood spot on filter paper
- Tandem Mass Spectrometry (modern method) - measures blood phenylalanine
- Done within 48-72 hours of birth (after protein feeding has begun)
- Normal blood phenylalanine: < 2 mg/dL; PKU: > 20 mg/dL (5-fold or more above normal)
Other Tests:
- Urine FeCl3 test - green color with phenylpyruvate (phenylketones)
- Serum phenylalanine level - elevated
- Molecular genetic testing - identifies specific PAH mutation (used for carrier testing)
Treatment
1. Dietary Restriction (Main treatment):
- Low-phenylalanine diet - restrict phenylalanine intake
- Avoid high-protein foods: meat, fish, dairy, eggs, nuts
- Use special phenylalanine-free amino acid formula
- Must be started early in life (within first few weeks of birth) - damage is irreversible
- Continue at least until adolescence; ideally lifelong
2. BH4 Supplementation:
- Sapropterin (synthetic BH4/tetrahydrobiopterin)
- Effective in variant PKU (BH4-deficient forms) and some classic PKU patients
- Cannot be used as sole therapy in classic PKU
3. Enzyme Substitution Therapy (Adults):
- Phenylalanine Ammonia-Lyase (PAL) - enzyme that metabolizes excess phenylalanine
- FDA-approved for adults with uncontrolled PKU
4. Gene Therapy:
- Under investigation
Maternal PKU (Important complication)
- Females with PKU who discontinue dietary treatment reach childbearing age with high phenylalanine levels
- Phenylalanine crosses the placenta and exerts teratogenic effects on the fetus
- Even though the infant is heterozygous (carrier), it is severely affected
- Outcomes: 75-90% of infants have intellectual disability, microcephaly, 15% have congenital heart disease
- This syndrome is called Maternal PKU syndrome
- Management: Maternal dietary restriction must be started before conception and continued throughout pregnancy
Summary Table (Quick Revision)
| Feature | Details |
|---|---|
| Enzyme deficient | Phenylalanine Hydroxylase (PAH) |
| Cofactor involved | Tetrahydrobiopterin (BH4) |
| Inheritance | Autosomal Recessive |
| Incidence | 1 in 10,000 |
| Key metabolite | Phenylpyruvate (in urine) |
| Characteristic smell | Musty/mousy odor |
| Screening test | Guthrie test / Tandem MS |
| Key clinical finding | Intellectual disability + hypopigmentation |
| Treatment | Low-phenylalanine diet |
Memory Tip (Mnemonics)
"PKU SMELLS":
- P - PAH enzyme deficient
- K - Ketones (phenylketones) in urine
- U - Urine smells musty
- S - Seizures
- M - Mental (intellectual) disability
- E - Eczema
- L - Light skin/hair
- L - Low tyrosine
- S - Screening (Guthrie test at birth)
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease | Lippincott Biochemistry 8th ed | Robbins & Kumar Basic Pathology
This is a shared conversation. Sign in to Orris to start your own chat.