Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Searching the Web
ARDS acute respiratory distress syndrome mechanism pathogenesis 2025
Reading File
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathogenesis mechanism alveolar injury
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content from Robbins Pathology, Murray & Nadel, and Fishman's, plus two excellent images. I have enough to compose a thorough answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is a syndrome of acute hypoxemic respiratory failure caused by diffuse alveolar injury. Its underlying basis is damage to both sides of the alveolar-capillary membrane — the endothelial lining of pulmonary capillaries and the epithelial lining of the alveolus — unleashing an amplifying inflammatory cascade.
The Central Mechanism Diagram
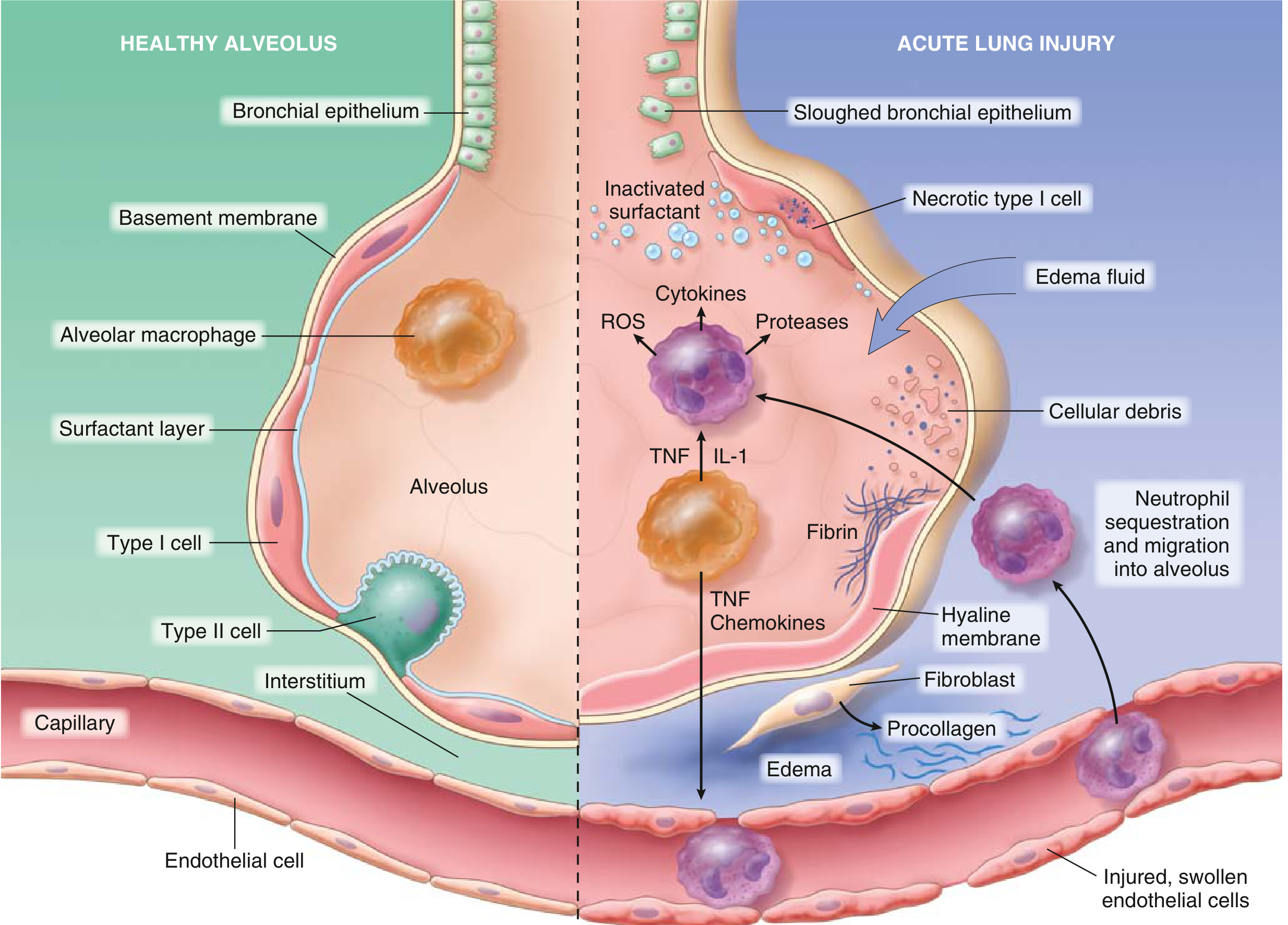
Fig. 11.2 from Robbins & Kumar Basic Pathology — healthy alveolus (left) vs. injured alveolus in ARDS (right)
Step-by-Step Pathogenesis
1. Initiating Insults
ARDS arises from direct (pulmonary) or indirect (systemic) insults to the lung:
| Direct | Indirect |
|---|---|
| Pneumonia (35–45%) | Sepsis (30–35%) |
| Aspiration | Severe pancreatitis |
| Inhalational injury | Major trauma / multiple fractures |
| Pulmonary contusion | Transfusion reactions (TRALI) |
| COVID-19 | Burns, hypotension |
2. Innate Immune Activation
The initiating insult triggers alveolar macrophages to release proinflammatory cytokines — primarily IL-1, TNF-α, chemokines (IL-8, CXCL1), and platelet-activating factor. These mediators:
- Activate pulmonary capillary endothelium (upregulating selectins and ICAM-1)
- Attract and activate neutrophils, which sequester in pulmonary capillaries
"Release of factors such as IL-1 and TNF leads to endothelial activation and sequestration and activation of neutrophils in pulmonary capillaries." — Robbins & Kumar Basic Pathology
3. Neutrophil-Mediated Injury (The Central Effector)
Activated neutrophils are the primary mediators of tissue destruction. They degranulate and release:
- Reactive oxygen species (ROS) — oxidative damage to endothelium and epithelium
- Proteases (elastase, collagenase, matrix metalloproteinases) — degrade basement membrane and structural proteins
- Leukotrienes and prostaglandins — amplify the inflammatory signal
- Additional cytokines — self-perpetuating loop
The balance between these destructive forces and endogenous counterparts (antiproteases like α₁-antitrypsin; antioxidants like superoxide dismutase) determines clinical severity.
4. Breakdown of the Alveolar-Capillary Barrier
Neutrophil-mediated injury causes:
Endothelial damage → increased vascular permeability → protein-rich fluid floods the interstitium and alveolar spaces → non-cardiogenic pulmonary edema
Type I pneumocyte necrosis → loss of the thin gas-exchange epithelium → impaired alveolar fluid clearance
Type II pneumocyte injury → reduced surfactant synthesis and secretion; phospholipase A₂ (from pancreatitis) enzymatically degrades existing surfactant → surfactant dysfunction and inactivation
5. Surfactant Loss and Alveolar Collapse
Surfactant normally reduces surface tension in alveoli, preventing collapse at end-expiration. Its inactivation causes:
- Diffuse alveolar collapse (atelectasis)
- Decreased lung compliance (stiff lungs)
- Intrapulmonary shunting → refractory hypoxemia
6. Coagulation Activation
Simultaneously, exposure of subendothelial collagen, release of tissue factor, and activation of platelets drives intravascular and intraalveolar fibrin deposition. This contributes to microvascular thrombosis (worsening perfusion) and formation of the pathognomonic hyaline membranes.
Histopathological Phases
The gross/microscopic picture of ARDS is diffuse alveolar damage (DAD), which evolves through three phases:
Phase 1: Exudative (Days 1–7)
- Lungs are dark red, firm, airless, and heavy
- Capillary congestion and interstitial edema
- Hyaline membranes lining alveolar ducts — fibrin-rich exudate + necrotic epithelial debris (the most characteristic finding)
- Neutrophilic infiltrates in capillaries and alveoli
Phase 2: Proliferative / Organizing (Days 7–21)
- Type II pneumocyte proliferation attempting to re-epithelialize the denuded alveolar surface
- Fibroblast infiltration, procollagen deposition
- Hyaline membranes begin to be resorbed or organized
Phase 3: Fibrotic (Beyond 3 weeks)
- Progressive alveolar septal fibrosis if repair fails
- Permanent architectural remodeling
- Reduced diffusion capacity and compliance
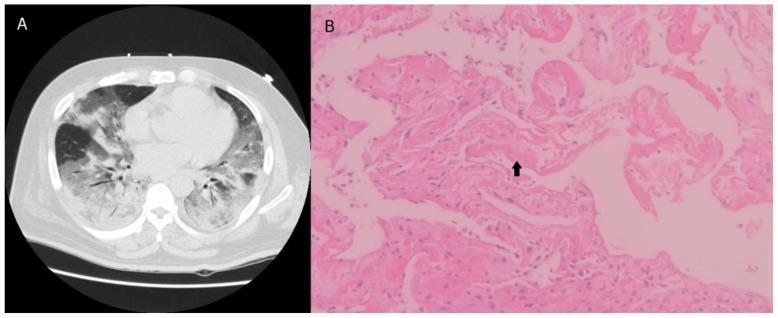
Left: Chest CT with bilateral ground-glass opacities in ARDS. Right: H&E histology showing hyaline membranes (arrow) and alveolar wall thickening — hallmarks of diffuse alveolar damage.
Physiological Consequences
| Mechanism | Physiological Effect |
|---|---|
| Alveolar flooding + atelectasis | ↓ FRC; intrapulmonary shunt → refractory hypoxemia |
| Surfactant loss | ↓ Lung compliance (stiff lungs) |
| Interstitial edema | Impaired O₂ diffusion |
| Microvascular thrombosis | ↑ Dead space, pulmonary hypertension |
| Widespread inflammation | Systemic SIRS → risk of multiorgan failure |
The hallmark clinical finding is a low PaO₂/FiO₂ ratio (Berlin definition: <300 mild, <200 moderate, <100 severe), reflecting the severity of ventilation-perfusion mismatch and shunt.
Key Cellular and Molecular Players Summary
| Player | Role |
|---|---|
| Macrophages | IL-1, TNF-α, chemokine release — initiate cascade |
| Neutrophils | ROS, proteases, cytokines — execute alveolar damage |
| Type I pneumocytes | Primary target of injury; necrosis destroys gas-exchange surface |
| Type II pneumocytes | Surfactant production; proliferate in repair phase |
| Endothelium | Permeability barrier; injury causes edema |
| Coagulation system | Fibrin deposition; hyaline membrane formation; microvascular occlusion |
| Fibroblasts | Procollagen → fibrosis in late phase |
Sources:
- Robbins & Kumar Basic Pathology, Chapter 11 (Acute Lung Injury and ARDS)
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter 134
- Fishman's Pulmonary Diseases and Disorders, Chapter on ARDS
- Sabiston Textbook of Surgery, Chapter on Critical Care
This is a shared conversation. Sign in to Orris to start your own chat.