Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathogenesis mechanism diagram
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Excellent content. I now have everything needed for a comprehensive explanation. Let me compile the full answer.
Acute Respiratory Distress Syndrome (ARDS) — Mechanism
ARDS is a syndrome of noncardiogenic pulmonary edema driven by severe diffuse alveolar damage (DAD). It is not a single pathophysiologic process but a collection of converging injury pathways that share a final common outcome: increased alveolar-capillary permeability, flooding of the air spaces, and refractory hypoxemia.
1. Triggers and Initiating Insults
ARDS arises from direct (pulmonary) or indirect (extrapulmonary) insults. The four conditions responsible for >50% of cases are:
| Direct Lung Injury | Indirect (Systemic) Injury |
|---|---|
| Pneumonia (viral, bacterial, Pneumocystis) | Sepsis (most common overall trigger) |
| Gastric aspiration | Severe trauma / head injury |
| Pulmonary contusion | Acute pancreatitis |
| Inhalational injury / smoke | Transfusion-related (TRALI) |
| Near-drowning | Burns, DIC |
— Robbins, Cotran & Kumar Pathologic Basis of Disease; Murray & Nadel's Textbook of Respiratory Medicine
2. Core Pathogenesis — The Alveolar-Capillary Barrier
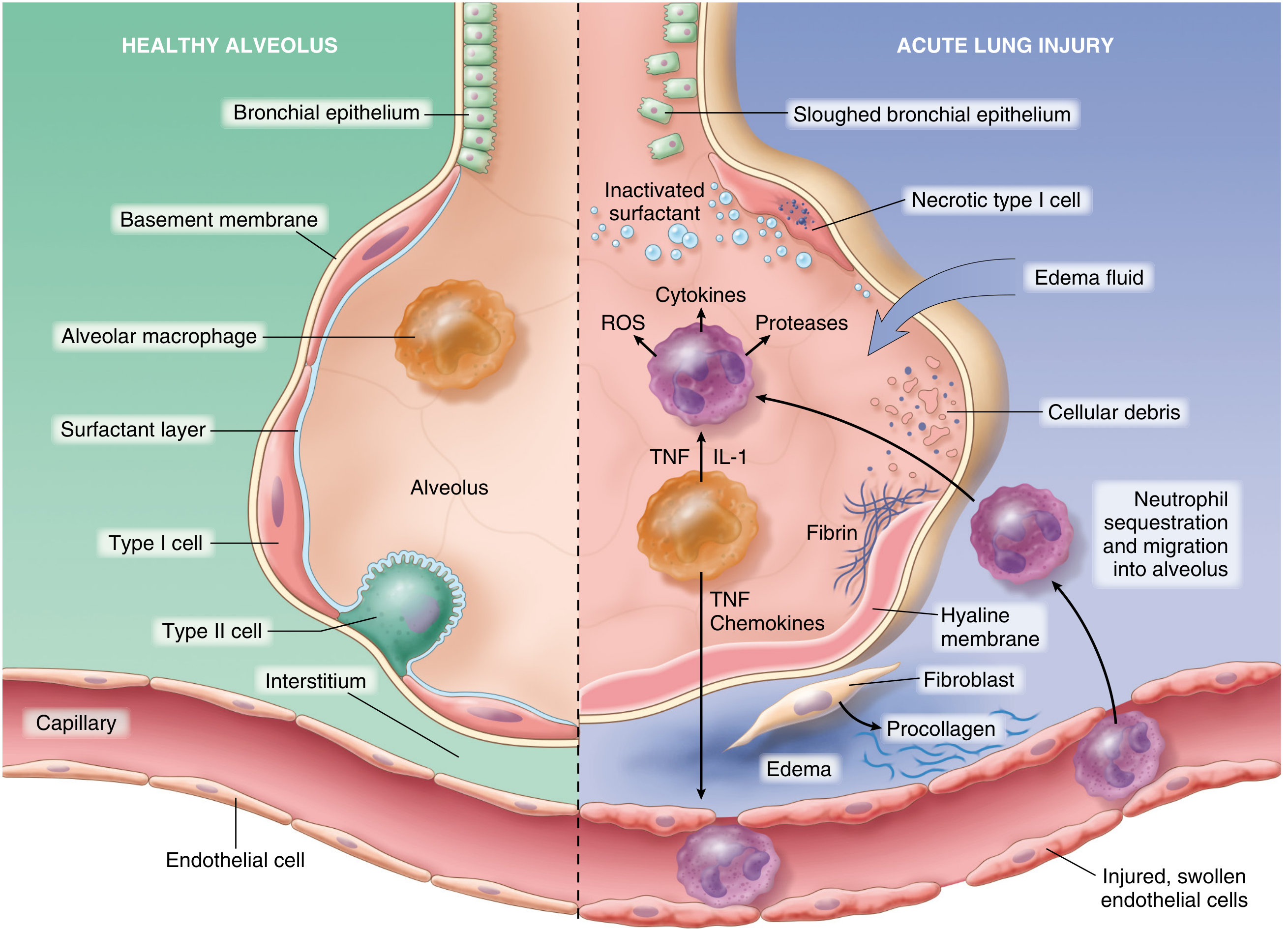
Fig. 15.3 — Normal alveolus (left) vs. acute lung injury (right). From Robbins, Cotran & Kumar Pathologic Basis of Disease.
Step-by-step sequence:
A. Endothelial Activation (Early Key Event)
- The trigger (sepsis, trauma, aspiration, etc.) produces either direct pneumocyte injury or circulating inflammatory mediators (LPS, cytokines, DAMPs).
- Resident alveolar macrophages sense pneumocyte injury and secrete TNF-α, IL-1β, IL-8, which act on the adjacent pulmonary microvascular endothelium.
- Alternatively, circulating mediators directly activate endothelial cells.
- Activated endothelium upregulates adhesion molecules (ICAM-1, E-selectin), procoagulant proteins, and chemokines.
B. Neutrophil Sequestration and Degranulation
- Neutrophils adhere to activated endothelium and migrate into the interstitium and alveoli.
- Once there, they degranulate, releasing:
- Proteases (elastase, metalloproteinases) — digest the basement membrane and extracellular matrix
- Reactive oxygen species (ROS) — directly injure pneumocytes and endothelial cells
- Cytokines — amplify the inflammatory loop
- Neutrophil extracellular traps (NETs) — contribute directly to lung tissue damage
C. Increased Alveolar-Capillary Permeability
- Injury to both endothelium (disrupting tight junctions) and alveolar epithelium (necrosis of type I pneumocytes) breaks down the alveolar-capillary membrane.
- Protein-rich, exudative fluid pours into the interstitium and then into alveolar spaces — noncardiogenic pulmonary edema.
- Unlike hydrostatic (cardiogenic) edema, this fluid has high protein content because the permeability barrier is structurally disrupted, not just pressure-overloaded.
D. Surfactant Dysfunction
- Type II pneumocytes (the source of surfactant) are damaged by:
- Direct injury from the insult
- Inflammatory mediators (phospholipase A₂ from activated macrophages/neutrophils degrades surfactant phospholipids)
- Inhibition by leaked plasma proteins in the alveolar fluid
- Loss of surfactant → increased alveolar surface tension → diffuse alveolar collapse (atelectasis) → worsening hypoxemia.
E. Coagulation Activation
- The inflammatory cascade activates the coagulation system within the alveolar space.
- Fibrin deposition in capillaries and alveoli contributes to microvascular thrombosis, impaired perfusion, and dead space.
- Together with cellular debris, this inspissated fibrin forms the pathognomonic hyaline membranes lining the alveolar walls.
F. The Inflammatory-Endothelial Feedback Loop
- Neutrophil-derived mediators cause further endothelial damage → more permeability → more neutrophil recruitment. This self-amplifying loop is the core engine of progressive ARDS.
- Angiopoietin-2 (a destabilizer of endothelial integrity) is elevated in ARDS and promotes vascular leak; Angiopoietin-1 (stabilizing) is reduced. This imbalance is an active research target.
— Murray & Nadel's Textbook of Respiratory Medicine, Chapter 134
3. Pathologic Phases (Diffuse Alveolar Damage — DAD)
DAD is the histologic hallmark of ARDS and progresses through three overlapping stages:
| Phase | Timing | Histology / Physiology |
|---|---|---|
| Exudative | Days 1–7 | Hyaline membranes, proteinaceous alveolar edema, neutrophilic infiltration, necrosis of type I pneumocytes |
| Proliferative | Days 7–21 | Hyaline membrane reorganization; type II pneumocyte hyperplasia; early fibrosis; decreased neutrophils |
| Fibrotic | >21 days (subset) | Collagen deposition, alveolar obliteration, pulmonary hypertension; impaired long-term function |
Note: only ~50% of ARDS patients show DAD on autopsy/biopsy. Those with confirmed DAD are younger, more severely ill, have worse compliance, and are ~5× more likely to die of hypoxic respiratory failure. — Murray & Nadel's, Chapter 134
4. Physiologic Consequences
| Mechanism | Effect |
|---|---|
| Alveolar flooding | ↓ Functional residual capacity (FRC) |
| Surfactant loss + alveolar collapse | ↓ Lung compliance (stiff lungs) |
| Flooded + collapsed alveoli still perfused | V/Q mismatch → right-to-left intrapulmonary shunt → refractory hypoxemia |
| Microvascular thrombosis + compression by PEEP | ↑ Dead space ventilation → ↑ PaCO₂ (or high minute ventilation needed to maintain normocapnia) |
| Hypoxic vasoconstriction + fibrin emboli | Pulmonary hypertension → right heart strain |
The hallmark is refractory hypoxemia — PaO₂/FiO₂ ratio <300 — that does not adequately respond to supplemental oxygen alone because the shunt fraction is fixed (collapsed/flooded alveoli accept oxygen from capillaries regardless of FiO₂).
5. Cellular Mechanisms Summary (Inflammatory Cascade)
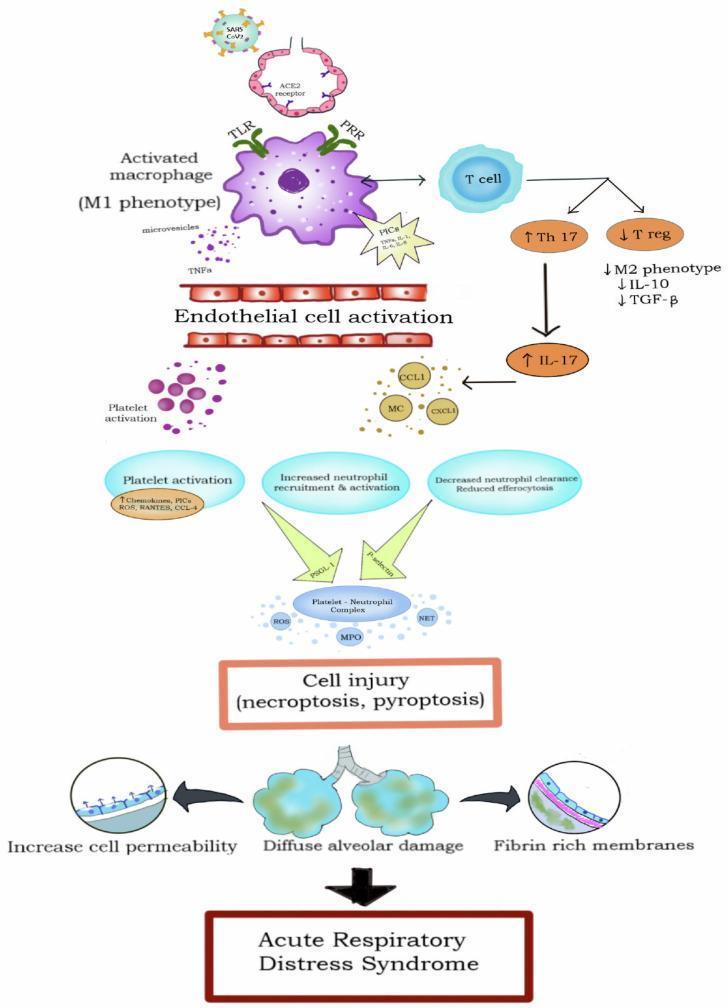
The cascade above illustrates the key cellular players:
- M1 macrophages (activated via TLR/PRR) → produce PICs (TNF-α, IL-1, IL-6)
- Endothelial cell activation → breakdown of vascular barrier
- Th17 upregulation / T-regulatory cell suppression → loss of anti-inflammatory balance (↓IL-10, ↓TGF-β)
- Platelet-neutrophil complexes → release ROS, myeloperoxidase (MPO), NETs → necroptosis/pyroptosis
- End result: increased cell permeability + diffuse alveolar damage + fibrin-rich hyaline membranes
6. Resolution vs. Fibrosis
If the inflammatory trigger abates:
- Macrophages clear alveolar debris and release TGF-β and PDGF → fibroblast proliferation and collagen synthesis.
- Type II pneumocytes proliferate to replace destroyed type I cells (type II → type I differentiation).
- Endothelial regeneration occurs from surviving capillary endothelium.
- In a subset, this fibroproliferative phase leads to permanent pulmonary fibrosis and long-term impairment.
7. Berlin Definition (Clinical Anchor)
| Criterion | Specification |
|---|---|
| Timing | Acute onset within 7 days of a known clinical insult |
| Imaging | Bilateral opacities on CXR/CT not fully explained by effusion, collapse, or nodules |
| Origin of edema | Not fully explained by cardiac failure or fluid overload |
| Oxygenation | Mild: P/F 200–300; Moderate: P/F 100–200; Severe: P/F <100 (with PEEP ≥5 cm H₂O) |
— Sabiston Textbook of Surgery; Murray & Nadel's Textbook of Respiratory Medicine
In summary, ARDS is fundamentally a disease of barrier failure: a systemic or local insult triggers macrophage activation and cytokine release, which recruits and activates neutrophils into the alveolar space. Neutrophil-derived oxidants, proteases, and NETs destroy the alveolar-capillary membrane, flooding alveoli with protein-rich fluid. Surfactant is inactivated, hyaline membranes form, and a self-amplifying inflammatory loop drives progressive lung stiffness, shunting, and refractory hypoxemia. The final clinical result is a lung that has lost its gas exchange capacity, requiring mechanical ventilatory support while the underlying insult is controlled.
This is a shared conversation. Sign in to Orris to start your own chat.