Metachromic leucodydtrophy
Reading File
Finding Sources
Finding Sources
Searching PubMed
metachromatic leukodystrophy
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive information from multiple authoritative textbooks. Here is a full synthesis:
Metachromatic Leukodystrophy (MLD)
Definition and Pathogenesis
Metachromatic leukodystrophy (MLD) is a lysosomal storage disorder (sphingolipidosis) caused by deficiency of the enzyme arylsulfatase A (ARSA), which normally converts sulfatide to cerebroside - a major component of myelin. Without ARSA, sulfatides accumulate in the CNS, peripheral nervous system, and other tissues (including the gallbladder), causing progressive demyelination.
A rarer form results from mutation in PSAP (encoding prosaposin), which is also needed for sulfatide breakdown.
- Inheritance: Autosomal recessive
- Gene locus: Chromosome 22q13 (ARSA gene); >60 mutations identified
- Stored substance: Sulfatides (galactosyl sulfatide)
- Sulfatides stain brown-orange (metachromatic) rather than purple with aniline dyes; also PAS-positive in frozen sections
Clinical Forms
| Form | Age of Onset | Predominant Features |
|---|---|---|
| Late infantile (most common) | 6 months - 2 years | Motor regression, gait disorder, hypotonia, lower-limb areflexia - peripheral neuropathy precedes CNS involvement |
| Juvenile | 3-16 years | Motor + behavioral problems, intellectual decline, ataxia, peripheral neuropathy |
| Adult | Mid-teens to 7th decade | Dementia, behavioral disturbances, psychiatric symptoms (hallucinations, delusions); often misdiagnosed as schizophrenia |
Late-Infantile Form (Classic):
- Progressive impairment of motor function (gait disorder, spasticity)
- Reduced speech output and mental regression
- Tendon reflexes initially brisk, later decreased/lost as peripheral nerves involved
- Later: visual impairment, strabismus, nystagmus, intention tremor, dysarthria, dysphagia, optic atrophy (1/3 of patients), macular gray degeneration
- Seizures are rare; no somatic abnormalities; head size usually normal
- Progresses to bedridden, quadriplegic, speechless state within 1-3 years
Adult Form:
- Often presents as psychiatric illness: auditory hallucinations, bizarre delusions, personality change, disinhibition, catatonic posturing
- Neurologic signs (dysarthria, spasticity) appear later
- Slowly evolving intellectual decline over 3-5 years
- Eventually progresses to loss of vision, speech, hearing, and decerebrate state
Pathology
- Widespread demyelination of cerebrum, cerebellum, spinal cord, and peripheral nerves
- Metachromatic granules in glial cells and engorged macrophages - characteristic finding
- Extensive segmental demyelination and abnormally thin myelin sheaths on nerve biopsy
- Metachromatic inclusions within Schwann cells and macrophages
MRI Findings
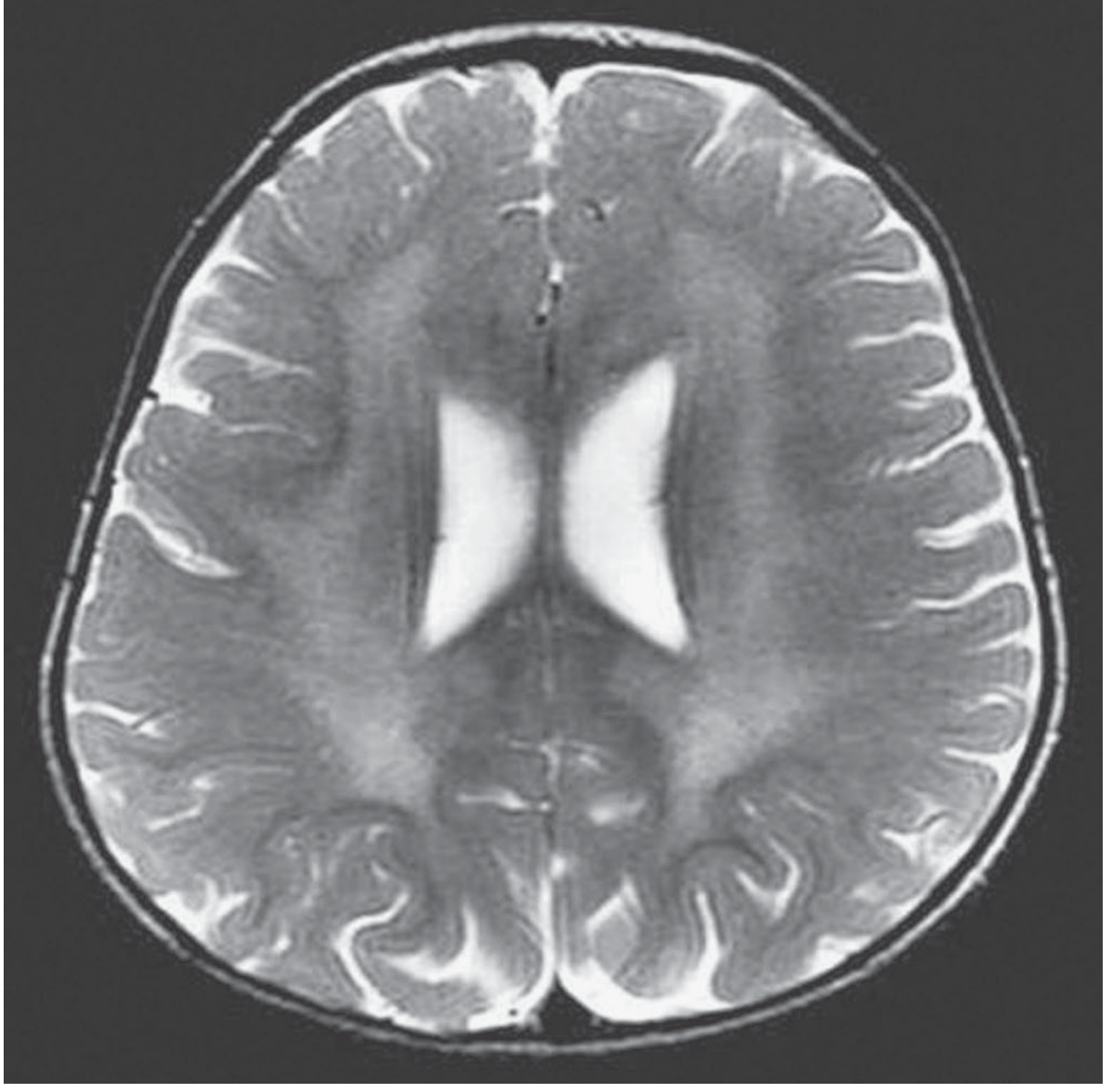
Axial T2-weighted MRI of a 2-year-old girl with MLD: symmetric central white matter hyperintensity with sparing of the subcortical arcuate (U) fibers. Confluent periventricular white matter lesions are hypointense on T1 and hyperintense on T2/FLAIR. - Adams and Victor's Neurology, 12th Ed.
Diagnosis
| Test | Finding |
|---|---|
| MRI brain | Symmetric confluent periventricular white matter hyperintensity (T2/FLAIR); sparing of subcortical U-fibers |
| CSF protein | Elevated: 75-250 mg/dL |
| Urine sulfatides | Markedly increased |
| ARSA enzyme activity | Absent/severely reduced in white blood cells, serum, cultured fibroblasts |
| Nerve conduction velocity | Markedly and uniformly slowed (especially late-infantile and juvenile forms) |
| Evoked potentials | Delayed visual and somatosensory evoked potential latencies (adult cases) |
| Peripheral nerve biopsy | Metachromatic inclusions in Schwann cells and macrophages (rarely needed now) |
| Molecular testing | ARSA gene mutation analysis (confirmatory) |
Important caveat - Pseudodeficiency: A pseudodeficiency allele (Pd) exists with carrier frequency of 15-20%. Homozygotes for this allele have enzyme activity ~10% of normal but NO clinical disease. Therefore, ARSA enzyme assay alone is not sufficient - must confirm with urine sulfatides AND/OR molecular testing, especially in atypical presentations.
Treatment
| Approach | Status |
|---|---|
| Allogeneic hematopoietic stem cell transplantation (HSCT) | Can slow progression if performed early in mildly affected patients; cord blood is a source; not curative |
| Gene therapy (ARSA gene) | Preliminary evidence for slowing progression; complications remain a concern |
| Gene therapy + HSCT combined | Under investigation |
| Enzyme replacement therapy (ERT) with recombinant human ARSA | Currently in clinical trials |
Key Points to Remember
- Enzyme: Arylsulfatase A (ARSA) deficiency - prevents sulfatide → cerebroside conversion
- Stored material: Sulfatides - stain metachromatic (brown-orange) with aniline dyes
- Inheritance: Autosomal recessive; gene on chromosome 22q13
- Classic finding: Symmetric periventricular white matter changes on MRI, with U-fiber sparing
- Peripheral neuropathy is an early feature (precedes CNS in infantile/juvenile forms)
- Adult form mimics psychiatric disease - a recognized cause of psychosis in adults
- Pseudodeficiency allele is common - always confirm with urine sulfatides or molecular testing
- Gallbladder involvement is common (hyperplastic polyps; possible increased carcinoma risk)
- CSF protein is consistently elevated
Sources: Adams and Victor's Principles of Neurology 12th Ed. | Goldman-Cecil Medicine | Bradley and Daroff's Neurology in Clinical Practice | Tietz Textbook of Laboratory Medicine 7th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.