Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND "pathophysiology"[MeSH Terms]
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathophysiology mechanism
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
I have comprehensive content from multiple authoritative sources. Let me now compile the full answer.
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
Definition
ARDS is a syndrome of acute respiratory failure characterized by noncardiogenic pulmonary edema due to diffuse alveolar damage (DAD), resulting in severe hypoxemia refractory to oxygen supplementation. The Berlin definition requires:
- Bilateral opacities on chest imaging
- PaO₂/FiO₂ ≤ 300 mmHg (on ≥5 cmH₂O PEEP)
- Onset within 1 week of a clinical insult
- Not fully explained by cardiac failure or fluid overload
Severity is graded: Mild (P/F 201–300), Moderate (101–200), Severe (≤100).
Precipitating Causes
| Direct Lung Injury | Indirect (Systemic) Injury |
|---|---|
| Pneumonia (35–45%) | Sepsis (30–35%) |
| Aspiration of gastric contents | Severe trauma / multiple fractures |
| Pulmonary contusion | Burns |
| Near-drowning | Pancreatitis |
| Toxic inhalation | Multiple transfusions (TRALI) |
| Drug overdose |
Core Pathogenesis
The underlying basis of ARDS is injury to both the alveolar epithelial lining and the pulmonary capillary endothelium — the alveolar-capillary membrane.
Step 1 — Initiating Injury
An insult (direct or systemic) triggers activation of alveolar macrophages, which release proinflammatory cytokines:
- IL-1β, IL-6, IL-8, TNF-α — amplify the inflammatory signal
- Leukotriene B4, platelet-activating factor — potent neutrophil chemoattractants
Step 2 — Neutrophil Sequestration and Activation
Cytokines (especially IL-8) cause sequestration of activated neutrophils in pulmonary capillaries, followed by transendothelial migration into the interstitium and alveolar spaces. Histologically, increased neutrophils appear within capillaries, the interstitium, and alveoli early in disease.
Activated neutrophils release:
- Reactive oxygen species (ROS) — oxidative damage to epithelial and endothelial cells
- Proteases (elastase, collagenase, MMPs) — degrade structural components of the alveolar wall
- More cytokines — sustain the inflammatory loop
The destructive forces can be counteracted by endogenous antiproteases and antioxidants — the balance between these determines clinical severity.
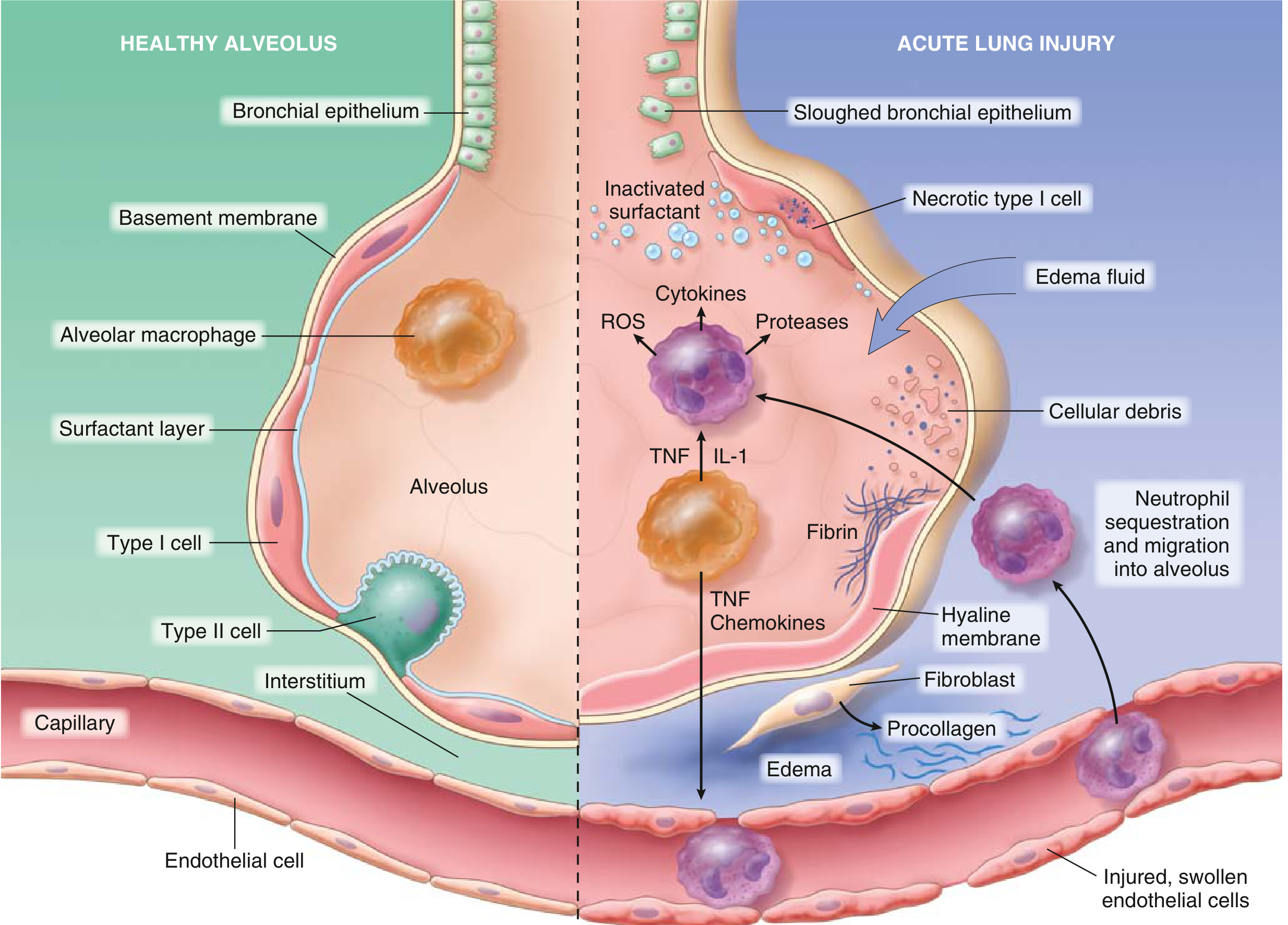
Step 3 — Breakdown of the Alveolar-Capillary Barrier
The assault on endothelium and epithelium causes:
- Endothelial injury → increased vascular permeability → protein-rich fluid floods the interstitium and alveoli
- Type I pneumocyte necrosis → loss of the tight epithelial barrier → fluid moves freely into alveolar spaces
- Microvascular thrombosis → microthrombi occlude pulmonary vessels, reducing blood flow to ventilated areas
Step 4 — Surfactant Dysfunction
- Plasma proteins and cellular debris flooding the alveolus inactivate surfactant
- Type II pneumocytes (which produce surfactant) are damaged
- Loss of surfactant → alveolar collapse (atelectasis), markedly decreased lung compliance
- Phospholipase A₂ (notably elevated in pancreatitis-associated ARDS) enzymatically degrades surfactant
Step 5 — Hyaline Membrane Formation
Fibrin-rich edema fluid mixes with cellular debris and dysfunctional surfactant to form hyaline membrane whorls lining distended alveolar ducts — the hallmark histologic lesion of DAD.
Pathophysiologic Consequences
| Derangement | Mechanism | Clinical Effect |
|---|---|---|
| Intrapulmonary shunt | Fluid-filled / collapsed alveoli perfused but not ventilated | Refractory hypoxemia (O₂ unresponsive) |
| Decreased compliance | Surfactant loss + edema + atelectasis | Stiff lungs, increased work of breathing |
| Increased dead space | Microvascular occlusion → ventilated areas not perfused | Hypercapnia, pulmonary hypertension |
| Pulmonary hypertension | Vasoconstriction (hypoxic) + microthrombi + fibrocellular proliferation | Increased RV afterload |
The dominant gas-exchange defect is large intrapulmonary shunt — this is why increasing FiO₂ alone has little effect. PEEP is the key intervention because it reopens collapsed alveoli, converting shunt back to normal V/Q.
Three Phases of ARDS (Clinical-Pathologic Course)
1. Exudative Phase (Days 1–7)
- Alveolar flooding with protein-rich edema, neutrophilic infiltration
- Hyaline membrane formation
- Type I pneumocyte necrosis
- Microthrombi in pulmonary vasculature
- Clinically: rapid-onset dyspnea, tachypnea, severe hypoxemia, bilateral opacities
2. Proliferative Phase (Days 7–21)
- Type II pneumocytes proliferate vigorously to regenerate the alveolar lining
- Fibrin-rich exudates begin to organize
- Interstitial inflammation and early fibrosis
- Most patients begin to recover; lung compliance may slowly improve
3. Fibrotic Phase (>3 weeks, a minority of patients)
- Extensive fibrosis and architectural destruction
- Bulla formation
- Reduced gas-exchange surface
- Persistent pulmonary hypertension
- High oxygen and ventilator dependency
Complete resolution occurs in a minority. More commonly, fibrin-rich exudates undergo organization, leading to fibrosis and alveolar septal thickening.
Ventilatior-Induced Lung Injury (VILI)
A critical secondary mechanism: mechanical ventilation itself can worsen ARDS through:
- Volutrauma — overdistension of aerated alveoli at high tidal volumes
- Atelectrauma — repeated collapse-reopening of unstable alveoli at low PEEP
- Biotrauma — cytokine/mediator release from mechanically stressed lung tissue causes systemic inflammation and distal organ dysfunction (ROS, leukotrienes, prostaglandins)
This is why lung-protective ventilation (6–8 mL/kg tidal volume, plateau pressure ≤30 cmH₂O, adequate PEEP) is the cornerstone of treatment.
Summary
ARDS is fundamentally a dysregulated inflammatory response at the alveolar-capillary interface. An initiating insult activates macrophages → cytokine storm → neutrophil recruitment and activation → oxidative/proteolytic damage to the alveolar-capillary barrier → flooding of alveoli with protein-rich fluid → surfactant inactivation → atelectasis, shunt, reduced compliance → profound refractory hypoxemia. Three phases (exudative, proliferative, fibrotic) describe the temporal evolution, with diffuse alveolar damage as the unifying histopathology.
Sources: Robbins & Kumar Basic Pathology; Harrison's Principles of Internal Medicine, 22e; Murray & Nadel's Textbook of Respiratory Medicine; Sabiston Textbook of Surgery
This is a shared conversation. Sign in to Orris to start your own chat.