I now have all the content I need to compose a comprehensive, exam-and-clinical-grade response. Let me deliver it.
The Floppy Infant — A Comprehensive Pediatric Master Guide
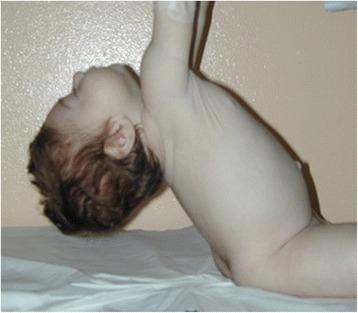
Pull-to-sit maneuver demonstrating severe head lag and axial hypotonia — the defining clinical sign of floppy infant syndrome.
1. Definition and Core Concepts
A floppy infant is one who exhibits hypotonia — defined as a decrease in resistance of muscle to passive stretch — either at birth or during early infancy. It is distinct from (but may coexist with):
- Weakness (decreased muscle power/force generation during voluntary contraction)
- Ligamentous laxity (increased joint range of motion without true neurological hypotonia)
Tone is an involuntary function maintained by the fusimotor system (muscle spindles via Ia afferent fibers + gamma motor neurons) and modulated at every level of the neuraxis — cortex, basal ganglia, cerebellum, brainstem, spinal cord, peripheral nerves, NMJ, and muscle. Disease at any of these levels can reduce tone.
Clinical pearl: Tone ≠ Strength. A hypotonic infant may have near-normal strength when aroused (e.g., during a blood draw), pointing toward a central rather than peripheral cause.
2. Pathophysiology: Central vs. Peripheral Hypotonia
The fundamental distinction that drives the entire diagnostic approach:
| Feature | Central Hypotonia | Peripheral Hypotonia |
|---|
| Level of lesion | Brain, brainstem, spinal cord | Motor neuron, peripheral nerve, NMJ, muscle |
| Prevalence | 60–80% of all cases | 15–30% of cases |
| Consciousness/alertness | Often encephalopathic, irritable, poor eye contact | Alert, awake, visually tracks normally |
| Deep tendon reflexes | Normal or brisk | Reduced or absent |
| Muscle strength | Relatively preserved (normal power with noxious stimuli) | Markedly reduced |
| Primitive reflexes | Abnormal (absent, obligatory, or exaggerated) | May be reduced or absent |
| Fasciculations | Absent | May be present (LMN disease) |
| Fisting (thumb adduction) | May be present | Absent |
| Seizures | May be present | Absent |
| Feeding difficulty | Due to encephalopathy | Due to bulbar weakness |
(Source: Bradley and Daroff's Neurology in Clinical Practice)
3. Etiology: Causes of the Floppy Infant Syndrome
CENTRAL CAUSES (Upper Motor Neuron / Brain)
A. Hypoxic-Ischemic Encephalopathy (HIE)
- Most common cause of neonatal hypotonia in the term infant
- Perinatal asphyxia → glutamate excitotoxicity → neuronal death
- Features: encephalopathy, seizures, abnormal tone, feeding difficulty
- Graded by Sarnat staging (mild/moderate/severe)
B. Chromosomal / Genetic Disorders
- Prader-Willi Syndrome (PWS): Chromosome 15q11-13 deletion (paternal) or maternal UPD; presents as the most classic "central floppy infant" — profound hypotonia, feeding difficulty requiring NG tube, later hyperphagia and obesity; cryptorchidism and almond-shaped eyes are clues
- Trisomy 21 (Down Syndrome): Hypotonia plus characteristic dysmorphic features; atlantoaxial instability requires screening
- Trisomies 13 and 18; various microdeletion syndromes
C. Structural Brain Malformations
- Lissencephaly, pachygyria, polymicrogyria, agenesis of corpus callosum
- Cerebellar hypoplasia/Dandy-Walker spectrum
- MRI brain essential for diagnosis
D. Metabolic / Inborn Errors of Metabolism
- Hypoglycemia, hypocalcemia, hypomagnesemia, hyperammonemia → all acutely reversible causes that must be excluded first
- Pompe disease (GSD type II / Acid Maltase Deficiency): Glycogen accumulation in muscle and anterior horn cells → profound weakness, cardiomegaly, hepatomegaly, macroglossia; CK elevated; treatment: alglucosidase alfa (ERT)
- Congenital Disorders of Glycosylation (CDG type Ia): Phosphomannomutase deficiency → hypotonia + hyporeflexia + cerebellar hypoplasia + inverted nipples + abnormal fat distribution; confirm with transferrin isoelectric focusing
- Mitochondrial disorders, organic acidemias, urea cycle defects
E. Peroxisomal Disorders
- Zellweger syndrome: Hypotonia + hepatomegaly + characteristic facies + absence of peroxisomes; elevated very-long-chain fatty acids (VLCFA)
PERIPHERAL CAUSES (Lower Motor Neuron)
A. Spinal Motor Neuron — Spinal Muscular Atrophy (SMA)
- Most important peripheral cause in a neonate/infant
- Loss of SMN1 gene (chromosome 5q) → anterior horn cell degeneration
- SMA Type 1 (Werdnig-Hoffmann disease): Onset < 6 months; never sits; paradoxical breathing (chest wall collapse with abdominal breathing); tongue fasciculations; absent DTRs; preserved alertness and eye movements; median survival < 2 years without treatment historically
- SMA Type 2: Onset 6–18 months; achieves sitting, never stands
- SMA Type 3 (Kugelberg-Welander): Onset after 18 months; ambulatory
B. Spinal Cord Injury
- Birth trauma (especially breech delivery, forceps delivery)
- Cervical spinal cord injury → generalized hypotonia below level of lesion
- May present with bladder distention, absent DTRs below level, associated HIE
C. Peripheral Nerve
- Congenital hypomyelinating neuropathy / Dejerine-Sottas disease: Absent DTRs, NCS shows severely reduced conduction velocities
- Hereditary motor and sensory neuropathies (HMSN)
D. Neuromuscular Junction (NMJ)
- Neonatal myasthenia gravis: Transient; maternal acetylcholine receptor antibodies cross placenta; improves within weeks; neostigmine test
- Congenital myasthenic syndromes: Permanent; due to mutations in NMJ proteins; edrophonium/neostigmine trial, repetitive nerve stimulation shows decrement
- Infant botulism: Clostridium botulinum spores (honey! soil) ingested → toxin blocks presynaptic ACh release; descending flaccid paralysis; constipation is often the first symptom; cranial nerve involvement (weak cry, poor suck, ptosis, dilated pupils); clear sensorium; EMG shows incremental response on high-frequency stimulation (SFEMG most sensitive); treatment = BabyBIG (human botulism immune globulin) — reduces hospitalization by 3–4 weeks
E. Muscle (Myopathies)
- Congenital myopathies (structural myopathies):
- Central core disease (RYR1 mutation): Hypotonia at birth, non-progressive; cores on Gomori trichrome; associated with malignant hyperthermia risk
- Nemaline (rod-body) myopathy: Rod-shaped inclusions on Gomori; can be fatal respiratory neonatal form
- Centronuclear/myotubular myopathy (MTM1 gene): Severe neonatal form in males (X-linked); central nuclei in myofibers; poor prognosis in X-linked form
- Fiber-type disproportion: Small type I fibers
- Congenital Muscular Dystrophies (CMD):
- Merosin-deficient CMD: Absent laminin-α2 on muscle biopsy; white matter changes on MRI; CMD confirmed by genetic testing
- Ullrich CMD, rigid spine syndrome:
- Myotonic Dystrophy type 1 (congenital form): Maternal CTG repeat expansion (DMPK gene); severe neonatal hypotonia, respiratory failure, bilateral facial weakness, talipes; mother often mildly symptomatic with grip myotonia → examine the mother!
- Infantile Pompe disease (also listed above as metabolic/combined)
4. Clinical Features and Physical Examination
Classic "Floppy" Postures on Inspection
- Supine resting posture: Legs in external rotation lying flat (frog-leg posture); arms extended by sides — contrast with the normal flexed frog posture of a healthy term infant
- Ventral suspension (prone suspension): Normal infant maintains head level with body and limbs semi-flexed; hypotonic infant drapes like an inverted "U" over the examiner's hand (rag-doll posture)
- Vertical suspension: Normal infant remains suspended with shoulder girdle strength; hypotonic infant slips through hands
- Pull-to-sit (traction response): Normal response includes elbow/knee/ankle flexion with minimal head lag (present normally after 33 weeks post-conceptional age); hypotonic infant shows profound head lag (see image above)
Key Clinical Signs to Differentiate Central from Peripheral
| Sign | Central | Peripheral |
|---|
| Alertness | Encephalopathic | Alert, bright-eyed |
| DTRs | Normal/brisk | Absent/reduced |
| Fasciculations | No | Tongue (SMA), limb (LMN) |
| Respiratory pattern | May have central apnea | Paradoxical chest movement (SMA) |
| Seizures | Yes | No |
| Dysmorphic features | Often (Down, PWS, brain malformations) | Rare (except CMD) |
| Ophthalmoplegia | Possible | Myasthenia, myotubular myopathy |
| Macroglossia | Pompe (also central) | — |
Infant botulism: profound generalized hypotonia with preserved alertness and descending paralysis pattern.
5. Diagnostic Approach
Step 1: Immediate Stabilization and Rule Out Reversible/Life-Threatening Causes
Any hypotonic infant must first be stabilized:
- Airway — hypotonia causes loss of airway control; may need intubation
- Breathing — paradoxical respiration suggests SMA or diaphragmatic weakness
- Circulation — sepsis is a reversible cause of hypotonia
Step 2: History (ESSENTIAL — Narrows Differential Before Any Test)
| Historical Feature | Implication |
|---|
| Reduced fetal movements | Peripheral/neuromuscular cause |
| Breech/difficult delivery, forceps | HIE, spinal cord injury |
| Maternal fever in pregnancy | In utero infection |
| Consanguinity | Autosomal recessive metabolic/genetic disorder |
| Maternal myotonia (grip myotonia on handshake) | Congenital myotonic dystrophy |
| Maternal acetylcholinesterase inhibitor use | Neonatal myasthenia |
| Honey exposure | Infant botulism |
| Older sibling with similar presentation | Genetic disorder |
| Maternal abortion history | Autosomal recessive lethals |
| Family history of muscle disease | CMD, SMA, myopathies |
Step 3: First-Line Investigations (All Hypotonic Infants)
| Investigation | Purpose |
|---|
| Blood glucose, Ca²⁺, Mg²⁺, Na⁺, K⁺ | Correct reversible metabolic causes immediately |
| Blood culture, CBC, CRP | Exclude sepsis |
| Ammonia | Urea cycle defects |
| Lactate, pyruvate | Mitochondrial disease, organic acidemias |
| LFTs, CK (creatine kinase) | Elevated in myopathies/CMD (CK > 1000 U/L suggests myopathy) |
| TFTs (thyroid function) | Hypothyroidism is a common, treatable cause |
| Urine drug screen | Neonatal opiate exposure |
| Urine organic acids, plasma amino acids | IEM screening |
| Chromosomes / chromosomal microarray | Down syndrome, PWS, chromosomal abnormalities |
Step 4: Targeted Second-Line Investigations Based on Clinical Localization
If CENTRAL suspected:
- MRI brain ± spine — structural malformation, HIE, leukodystrophy, Dandy-Walker
- EEG — subclinical seizures
- TORCH serology
- Chromosomal microarray / exome sequencing
- Urine/serum for CDG (transferrin isoelectric focusing)
- Very-long-chain fatty acids (Zellweger, peroxisomal)
- Plasma amino acids, urine organic acids, acylcarnitine profile
If PERIPHERAL suspected:
- Nerve conduction studies (NCS) + EMG
- Decreased conduction velocity → neuropathy
- Absent sensory potentials → neuropathy
- Fibrillations + positive sharp waves → denervation (SMA)
- Decremental response on repetitive stimulation → NMJ (myasthenia)
- Incremental response at high frequency → botulism
- CK: Markedly elevated in congenital muscular dystrophies
- SMN1 gene deletion analysis — now first-line for suspected SMA (detects 95–98% of SMA)
- Anti-AChR antibodies — neonatal myasthenia
- Stool culture for C. botulinum and toxin — infant botulism
- Muscle biopsy (histochemistry, electron microscopy) — congenital myopathy diagnosis
- Skin/peripheral nerve biopsy — infantile neuroaxonal dystrophy
- Genetic panel / whole exome sequencing — where molecular diagnosis needed
Hammersmith Infant Neurological Examination (HINE): The strongest validated tool for quantifying hypotonia severity in infants 2 months to 2 years (Hidalgo Robles et al., 2024, PMID 38391868). Use it for serial assessments.
6. Red Flags — Do Not Miss
| Red Flag | Action |
|---|
| Respiratory failure — paradoxical breathing, SpO₂ falling | Immediate ICU; ventilatory support; suspect SMA type 1 |
| No spontaneous movements + absent reflexes | SMA type 1 or spinal cord injury — urgent SMN1 deletion analysis |
| Macroglossia + cardiomegaly | Pompe disease — urgent cardiac echo + acid alpha-glucosidase assay |
| Constipation + descending weakness + afebrile | Infant botulism — stool C. botulinum; administer BabyBIG |
| Profound hypotonia in son of mildly affected mother | Congenital myotonic dystrophy — DM1 gene repeat analysis in mother |
| Consanguinity + metabolic derangement | IEM — urgent metabolic workup |
| Encephalopathy + seizures + dysmorphic | Brain MRI; chromosomal microarray; consider HIE protocol (therapeutic hypothermia if criteria met) |
| Inverted nipples + cerebellar hypoplasia on MRI | CDG syndrome — transferrin isoelectric focusing |
7. Management
A. Acute / Emergency Management
- Stabilize the airway — anticipate need for intubation in any infant with respiratory distress; diaphragm weakness means rapid deterioration
- Correct reversible causes immediately:
- Hypoglycemia → IV dextrose
- Hypocalcemia → IV calcium gluconate
- Hypothyroidism → levothyroxine
- Sepsis → broad-spectrum antibiotics
- Therapeutic hypothermia — if HIE is the cause, initiate within 6 hours of birth (33–34°C for 72 hours) per standard AAP/WHO protocols (must meet Sarnat grade II–III criteria)
- BabyBIG (Human Botulism Immune Globulin) — administer IV as early as possible in infant botulism; reduces hospitalization by average 3.6 weeks; FDA-approved; do NOT give aminoglycosides (potentiate NMJ blockade)
B. Disease-Specific Pharmacological Treatment
| Condition | Treatment |
|---|
| SMA type 1 | Nusinersen (Spinraza) — intrathecal antisense oligonucleotide, FDA-approved; OR Onasemnogene abeparvovec (Zolgensma) — one-time IV gene therapy (SMN1 gene replacement); Risdiplam (Evrysdi) — oral SMN2 splicing modifier. Pre-symptomatic treatment (via newborn screening) dramatically improves outcomes |
| Pompe disease | Alglucosidase alfa (Myozyme) — IV enzyme replacement therapy; initiate as early as possible; reduces cardiac mass and prolongs ventilator-free survival |
| Congenital hypothyroidism | Levothyroxine — treat immediately to prevent irreversible neurodevelopmental damage |
| Neonatal myasthenia | Neostigmine / pyridostigmine; self-limiting within weeks as maternal antibodies wane |
| Infant botulism | BabyBIG (California Infant Botulism Treatment & Prevention Program) |
| Myotonic dystrophy | Supportive; mexiletine for myotonia (not curative); multidisciplinary |
| HIE | Therapeutic hypothermia (if criteria); seizure management; neuroprotection |
C. Non-Pharmacological Management (Critical in ALL floppy infants)
- Physiotherapy — prevent contractures, promote motor development; stretching and positioning from early infancy
- Occupational therapy — activities of daily living, adaptive equipment
- Speech and feeding therapy — coordinate suck-swallow-breathe; assess for aspiration risk
- Nasogastric tube (NG) or gastrostomy (PEG/G-tube) — if feeding inadequate or aspiration risk high
- Non-invasive ventilation (NIV/BiPAP) — for chronic respiratory insufficiency (SMA type 1, CMD)
- Orthopedic management — scoliosis surveillance and bracing/surgery; joint contracture management
- Aggressive respiratory infection management + annual influenza vaccination
- Cardiac surveillance — echo in Pompe, congenital myotonic dystrophy, Emery-Dreifuss
- GERD management — common in hypotonic infants
8. Differential Diagnosis Summary (Practical Framework)
FLOPPY INFANT
│
├── ALERT, NORMAL REFLEXES, POOR POWER
│ └── PERIPHERAL CAUSE
│ ├── Absent DTRs + fasciculations → SMA (SMN1 analysis URGENT)
│ ├── Absent DTRs, no fasciculations, NCS abnormal → Neuropathy
│ ├── Ptosis + fatigability, decrement on RNS → Myasthenia
│ ├── Constipation + descending weakness + afebrile → Botulism
│ └── Elevated CK + muscle biopsy changes → CMD / Congenital myopathy
│
└── ENCEPHALOPATHIC, NORMAL/BRISK REFLEXES, RELATIVELY NORMAL POWER
└── CENTRAL CAUSE
├── Birth asphyxia + seizures → HIE
├── Dysmorphic + chromosomal → Trisomy 21/13/18, PWS, microdeletion
├── Macroglossia + cardiomegaly → Pompe
├── Constipation + later hyperphagia + genital hypoplasia → PWS
├── MRI white matter changes → Leukodystrophy, CDG, lysosomal
└── Normal MRI + normal chromosomes → Benign congenital hypotonia
9. Complications and Prevention
| Complication | Prevention/Management |
|---|
| Respiratory failure | Early NIV/intubation; NMJ disease needs serial FVC monitoring; SMA-specific interventions |
| Aspiration pneumonia | Feeding therapy; thickened feeds; G-tube if indicated; semi-upright positioning |
| Scoliosis | Serial spinal X-rays; early bracing; spinal fusion when >40–50° |
| Joint contractures | Early physiotherapy; serial casting; surgery as needed |
| Failure to thrive | Nutritional support; caloric enrichment; NG/G-tube |
| Developmental delay | Early intervention programs (EI); speech, OT, PT; individualized education plans (IEP) |
| Malignant hyperthermia (central core disease/RYR1) | Alert anesthesia team; avoid triggering agents (succinylcholine, volatile anesthetics) |
| Cardiac arrhythmia/failure | Echo surveillance; cardiology in Pompe, myotonic dystrophy, Emery-Dreifuss |
10. Prognosis and Long-Term Outcomes
| Etiology | Prognosis |
|---|
| HIE (mild) | Good neurological outcome; 95%+ normal |
| HIE (severe) | High mortality; significant neurodevelopmental morbidity (CP, epilepsy, ID) |
| Down Syndrome | Variable; early intervention critical; life expectancy into 50–60s now |
| Prader-Willi Syndrome | Hypotonia improves with age; later behavioral and metabolic issues; growth hormone therapy helps |
| SMA type 1 (untreated) | Median survival < 2 years; death from respiratory failure |
| SMA type 1 (with gene therapy — Zolgensma) | Dramatic improvement; many children achieve motor milestones never seen before; trials show 100% survival at 14 months in pre-symptomatic treatment |
| Infant botulism | With BabyBIG: excellent; full recovery expected; mortality < 1% |
| Pompe disease | ERT improves survival significantly; outcome depends on genotype (infantile vs. late-onset) and timing of treatment initiation |
| Congenital myotonic dystrophy | Neonatal form has high early mortality (respiratory); survivors have intellectual disability |
| Central core myopathy | Usually non-progressive or slowly progressive; most achieve ambulation |
| Benign congenital hypotonia | By definition, improves with age; good long-term prognosis |
11. Long-Term Follow-Up
Conditions requiring structured lifelong follow-up:
SMA:
- Serial neurological assessments; respiratory function (FVC); motor milestone tracking
- Repeat nusinersen injections (loading: 4 doses over 63 days; maintenance: every 4 months)
- Scoliosis, nutritional, and swallowing surveillance
Pompe Disease:
- ERT infusions every 2 weeks lifelong
- Cardiac echo (infantile form has cardiomyopathy)
- Respiratory function; GI; audiology
Down Syndrome:
- Echocardiogram at birth (AVSD, VSD)
- Thyroid function annually
- Atlantoaxial instability screening at 3–5 years
- Audiology, ophthalmology annually
- Sleep study for OSA
- Leukemia surveillance
Prader-Willi Syndrome:
- Growth hormone therapy from infancy (FDA-approved)
- Endocrinology (hypogonadism, diabetes risk)
- Behavioral/psychiatric support
- Obesity prevention (strict dietary environment)
- GnRH analogs for puberty management
Congenital Muscular Dystrophies:
- Cardiac MRI/echo annually
- Pulmonary function + sleep study
- Orthopedic: scoliosis, contractures
- Nutritional assessment
12. Patient-Centered and Family-Centered Care
Psychological Impact on Families
Receiving a diagnosis of a chronic neuromuscular condition is catastrophically distressing. Key principles:
- Break the news honestly and compassionately — use clear, non-medical language; involve both parents and key family members
- Avoid false reassurance — but maintain hope around disease-modifying treatments (especially SMA gene therapy)
- Acknowledge grief — anticipatory grief for a future the family imagined; normalize this response
- Provide written information and direct to reputable disease-specific organizations (Cure SMA, NORD, Prader-Willi Syndrome Association)
Multidisciplinary Team (MDT) — Essential
A floppy infant with a neuromuscular diagnosis requires all of:
- Pediatric neurologist
- Pulmonologist / respiratory therapist
- Gastroenterologist / dietitian
- Physiotherapist (from day 1)
- Occupational therapist
- Speech and language therapist / feeding specialist
- Cardiologist (selected diagnoses)
- Orthopedic surgeon (scoliosis, contractures)
- Geneticist (for family counseling, recurrence risk)
- Palliative care team (when prognosis is guarded)
- Social worker and psychologist
Ethical Considerations
- Informed consent for genetic testing — implications for siblings and parents (carrier status)
- Newborn screening for SMA — now on the US Recommended Uniform Screening Panel (RUSP); enables pre-symptomatic treatment with dramatically superior outcomes; but raises questions about parental anxiety with early disclosure
- Goals of care discussions — particularly in SMA type 1 and severe CMD; ventilatory support decisions should be made collaboratively with family, guided by the child's best interests
- Transition to adult care — plan early for adolescents with chronic neuromuscular conditions
13. Clinical Reasoning Summary
When you see a floppy infant at the bedside, the mental algorithm is:
- Stabilize first — airway, breathing, circulation, glucose, calcium
- Examine — is the baby alert with absent reflexes (peripheral) or encephalopathic with normal reflexes (central)?
- History focus — maternal, family, delivery, exposures (honey!)
- First-line labs — rule out reversible: glucose, calcium, TFTs, sepsis screen, CK, ammonia, lactate
- Central → MRI brain; Peripheral → NCS/EMG → targeted genetic testing
- SMA is the most dangerous peripheral cause you must not miss — send SMN1 deletion analysis early; time to treatment is critical
- Infant botulism must be recognized clinically — stool test takes days; give BabyBIG on clinical suspicion if descending paralysis + constipation + afebrile + clear sensorium
- Pompe disease must be recognized early — cardiomegaly + macroglossia + weakness = send acid alpha-glucosidase before the heart fails
Key References
- Bradley and Daroff's Neurology in Clinical Practice — Chapter 30: The Floppy Infant
- Adams and Victor's Principles of Neurology, 12th ed. — Chapter 37: Flaccid Paralyses and the Floppy Infant Syndrome (Table 37-7)
- Harrison's Principles of Internal Medicine 22E (2025) — Lower Motor Neuron Disorders, SMA
- Rosen's Emergency Medicine — Infant Botulism (Chapter 169)
- Hidalgo Robles Á et al. "Identifying and Evaluating Young Children with Developmental Central Hypotonia." Healthcare (Basel) 2024. PMID 38391868 — Validates HINE as the strongest assessment tool for central hypotonia
- Francisco R et al. "Neonatal tone management." Semin Fetal Neonatal Med 2024. PMID 39537451 — MDT approach, importance of serial examinations and early intervention
- AAP Newborn Screening — SMA added to RUSP 2018
- Stonybrook/Pediatrics in Review 2009: "The Floppy Infant: Evaluation of Hypotonia" — systematic approach framework still widely used
This covers the full scope of the floppy infant from first principles to bedside to long-term follow-up. When you encounter this clinically, the single most important action after stabilization is localizing the lesion (central vs. peripheral) through the history and examination — because that branch point determines every subsequent investigation and intervention.