Routes of drug administration
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a thorough, well-illustrated answer. Here it is:
Routes of Drug Administration
The route of administration is determined by the drug's physicochemical properties (lipid/water solubility, ionization, molecular size) and the therapeutic goal (speed of onset, local vs systemic effect, patient status). Routes fall into three major categories: enteral, parenteral, and other (miscellaneous).
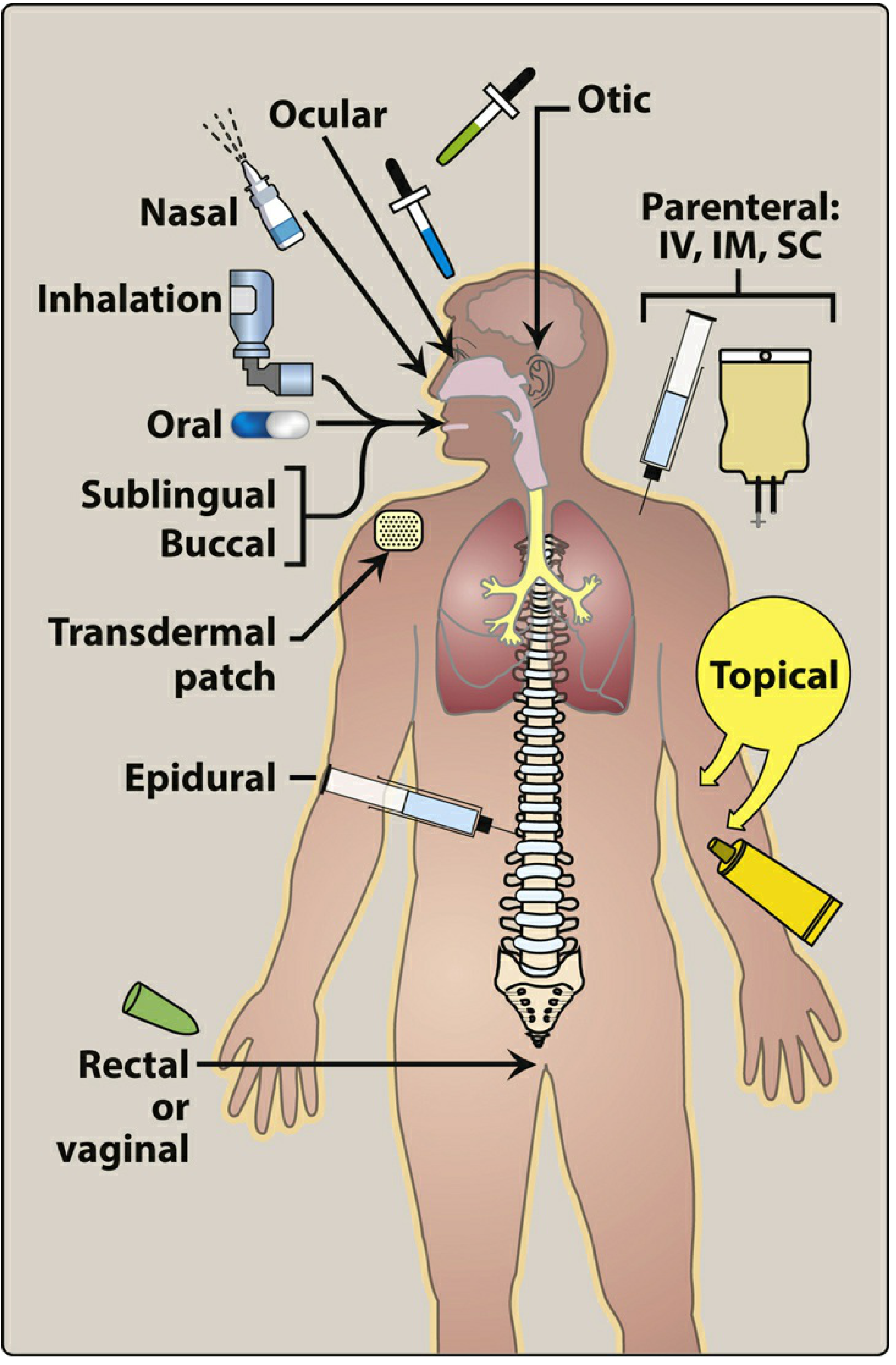
1. Enteral Routes
Enteral administration involves delivery of drug via the GI tract.
A. Oral
The most common, convenient, and economical route. The drug is swallowed and absorbed primarily from the small intestine (surface area ~200 m²).
Advantages:
- Easy self-administration
- Safe - toxicity/overdose can be managed with activated charcoal
- Wide range of formulations available
Disadvantages:
- Variable and potentially erratic absorption (bioavailability F = 0.05 to 1)
- Low gastric pH destroys acid-labile drugs (e.g., insulin, penicillin G)
- Subject to first-pass hepatic metabolism (reduces systemic availability)
- Requires patient cooperation
- Food, GI motility, and gut microbiome can all alter absorption
- Inappropriate for unconscious or vomiting patients
Special formulations:
- Enteric-coated preparations (e.g., omeprazole, aspirin): a chemical envelope prevents dissolution in the stomach, protecting acid-labile drugs or preventing gastric irritation
- Extended-release (ER/XR/XL/SR/CR): controlled coatings slow drug release, prolong duration of action, reduce dosing frequency, and smooth out peak/trough fluctuations. Useful for drugs with short half-lives (e.g., ER morphine reduces dosing from 6x/day to 2x/day)
Examples: Acetaminophen, amoxicillin
B. Sublingual and Buccal
- Sublingual: drug placed under the tongue
- Buccal: drug placed between the cheek and gum
Both bypass the GI environment and first-pass metabolism via direct absorption into systemic venous drainage. The neutral pH of saliva also maintains drug stability.
Advantages: Rapid onset, bypass of first-pass effect, avoid gastric acid destruction
Disadvantages: Limited to small doses; only certain drugs have adequate absorption; drug may be partially swallowed
Examples: Nitroglycerin (SL) for angina, buprenorphine
C. Rectal
About 50% of rectal venous drainage bypasses the portal circulation, so first-pass metabolism is partially avoided. Useful when the patient is vomiting, unconscious, or cannot take oral medications.
Disadvantages: Absorption is often erratic and incomplete; many drugs irritate the rectal mucosa; patient compliance is low.
2. Parenteral Routes
Parenteral administration introduces drug directly into the systemic circulation, bypassing the GI tract entirely. It is used for:
- Drugs poorly absorbed or inactivated in the GI tract (e.g., heparin, insulin)
- Unconscious patients
- Situations requiring rapid onset or precise dose control
General disadvantages: Irreversible once administered; risk of pain, infection, local tissue damage, hemolysis, thrombophlebitis; requires trained personnel and sterile technique.
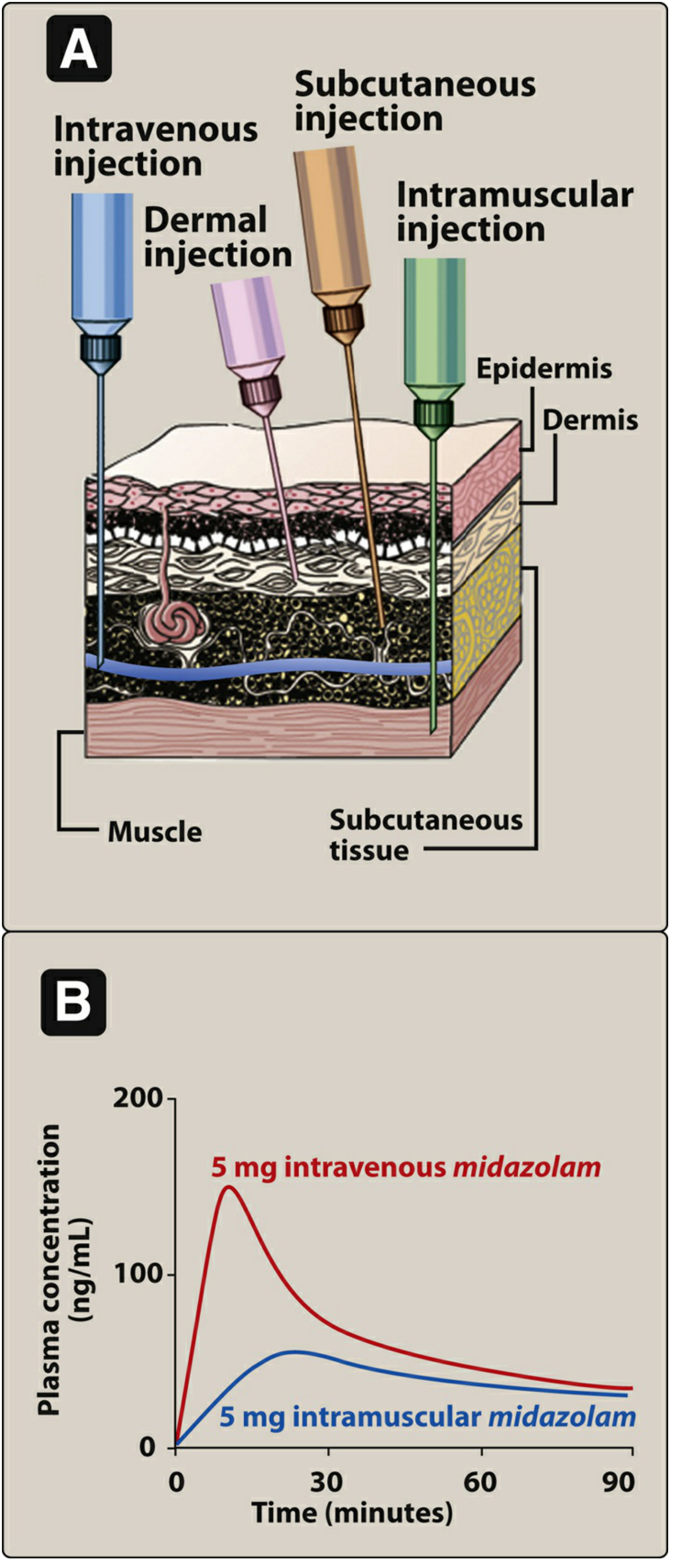
A. Intravenous (IV)
Bioavailability: F = 1 (by definition) - absorption is circumvented entirely.
- Most common parenteral route
- Bolus injection: full dose reaches systemic circulation almost immediately
- IV infusion: slower delivery, lower peak concentration, longer effect
- Permits titration of dosage; ideal in emergencies
- Required for high-molecular-weight proteins/peptides (e.g., monoclonal antibodies)
- Can deliver large volumes and irritating substances when diluted
Limitations: Increased risk of adverse effects; solutions must be injected slowly; oily or poorly soluble substances cannot be given IV.
Examples: Rocuronium, vancomycin, heparin
B. Intramuscular (IM)
Bioavailability: F = 0.75 to 1
- Aqueous solutions are absorbed rapidly; depot preparations (in oily vehicles like polyethylene glycol) are absorbed slowly and provide sustained release as the vehicle diffuses out
- Suitable for moderate volumes
- Can accept oily vehicles and some irritating substances
Limitations: Pain or necrosis from irritating substances; contraindicated during anticoagulant therapy (risk of hematoma); can falsely elevate creatine kinase.
Examples: Haloperidol decanoate (depot), penicillin G benzathine, IM vaccines
C. Subcutaneous (SC)
Bioavailability: F = 0.75 to 1
- Slower absorption than IM (via simple diffusion); slower than IV
- Provides constant, slow, sustained effects
- Minimizes hemolysis and thrombosis risk
- Not suitable for drugs that cause tissue irritation (risk of pain and necrosis)
- Can be used for slow-release implants
Examples: Insulin, low-molecular-weight heparin, epinephrine auto-injectors
D. Intradermal (ID)
- Injection into the dermis (vascular layer beneath the epidermis)
- Used for diagnostic tests and desensitization procedures
Examples: Tuberculin skin test (Mantoux), allergy testing, BCG vaccine
3. Other Routes
A. Inhalation
Rapid delivery across the large mucosal surface area of the respiratory tract and pulmonary epithelium - onset nearly as fast as IV bolus.
- Gases (e.g., volatile anesthetics) and aerosol preparations
- Drug deposited directly at the site of action in pulmonary disease, minimizing systemic side effects
- Nasal route targets the nasal mucosa for local effect (e.g., allergic rhinitis)
Examples: Salbutamol (asthma), inhaled corticosteroids, nitrous oxide, intranasal fluticasone
B. Transdermal
Systemic drug delivery through the skin, usually via a patch. Absorption rate depends on skin characteristics at the application site and the lipid solubility of the drug.
- Provides slow, sustained, continuous drug delivery
- Bypasses first-pass metabolism
- Non-invasive
Examples: Nicotine patch, fentanyl patch, nitroglycerin patch, estrogen patch
C. Intrathecal / Intraventricular
The blood-brain barrier (BBB) limits CNS drug penetration. When rapid CNS effects are needed, drugs are injected directly into the subarachnoid space (intrathecal) or cerebral ventricles.
Examples: Spinal anesthesia (bupivacaine), intrathecal methotrexate for CNS lymphoma, intrathecal baclofen for spasticity
D. Topical
Local application to a surface (skin, eye, ear, mucous membranes) for a local effect, avoiding systemic absorption.
Examples: Topical corticosteroids, ophthalmic drops, otic drops, antifungal creams
E. Epidural
Injection into the epidural space (outside the dura mater) provides regional analgesia/anesthesia.
Examples: Epidural bupivacaine/fentanyl in obstetrics, postoperative pain management
F. Rectal / Vaginal
Already mentioned under enteral (rectal). The vaginal route is used for local or systemic hormonal effects.
Examples: Vaginal progesterone gel, misoprostol, antifungal suppositories
Summary Table (Goodman & Gilman's Table 2-1)
| Route | Bioavailability (F) | Onset | Key Advantage | Key Limitation |
|---|---|---|---|---|
| Intravenous | 1.0 (by definition) | Immediate | Emergency use; exact dose control | Adverse effects; irreversible |
| Intramuscular | 0.75-1.0 | Rapid (aqueous) / Slow (depot) | Depot preparations possible | Pain; avoid with anticoagulants |
| Subcutaneous | 0.75-1.0 | Slow, sustained | Implants; constant release | Not for irritating drugs |
| Oral | 0.05-1.0 | Variable | Safest; most convenient; cheapest | First-pass effect; GI destruction |
| Sublingual/Buccal | High for some drugs | Rapid | Bypasses first-pass; rapid onset | Small doses only; limited drugs |
| Inhalation | High (pulmonary) | Very rapid | Local delivery to lungs | Technique-dependent |
| Transdermal | Moderate-high | Slow | Sustained; bypasses first-pass | Limited by skin permeability |
| Rectal | Partial (50% bypass) | Moderate | Useful in vomiting/unconscious | Erratic absorption; inconvenient |
| Intrathecal | N/A (direct CNS) | Rapid (CNS) | Overcomes BBB | Technically demanding; risky |
Sources:
- Lippincott Illustrated Reviews: Pharmacology, Chapter 1 (Routes of Drug Administration)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, Chapter 2 (Drug Absorption, Bioavailability, and Routes of Administration, Table 2-1)
Note on drug absorption and factors influencing it
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have everything needed for a thorough, well-illustrated note on drug absorption. Here it is:
Drug Absorption and Factors Influencing It
Definition
Absorption is the transfer of a drug from its site of administration into the bloodstream (systemic circulation). The rate and extent of absorption depend on the environment at the absorption site, the physicochemical characteristics of the drug, and the route of administration. Routes other than intravenous may result in partial absorption and lower bioavailability.
Mechanisms of Drug Absorption
Drugs cross cell membranes via four main mechanisms:
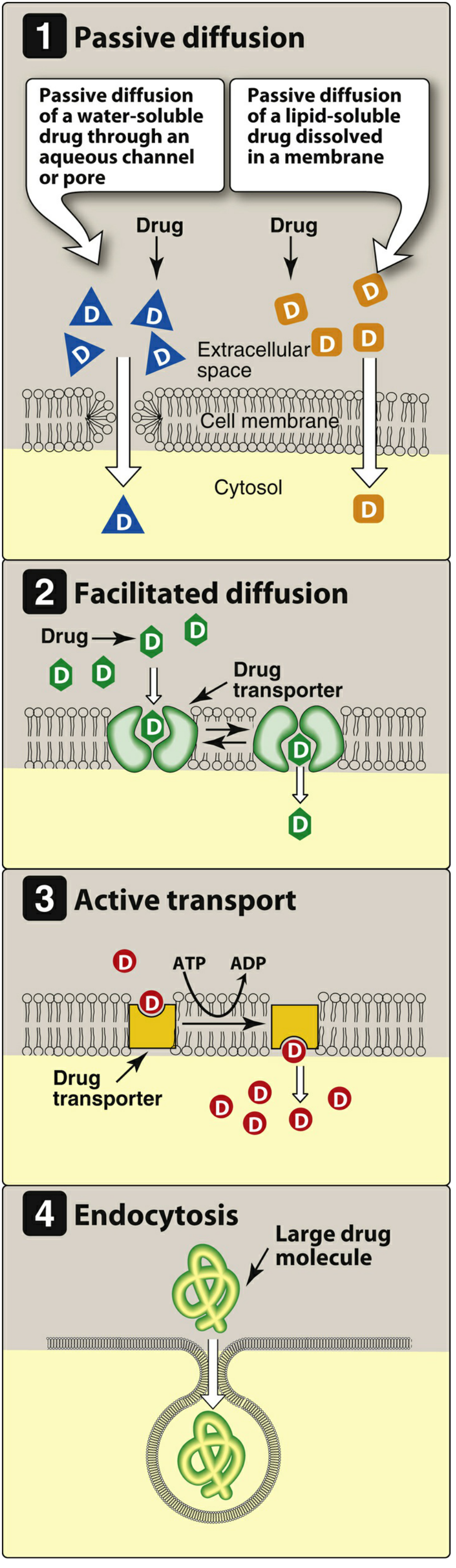
1. Passive Diffusion
The most common mechanism for the vast majority of drugs. Movement is driven by the concentration gradient - drug moves from high to low concentration.
- Requires no carrier protein, no energy
- Not saturable; shows low structural specificity
- Water-soluble drugs pass through aqueous channels/pores
- Lipid-soluble drugs dissolve directly into the lipid bilayer and cross readily
2. Facilitated Diffusion
Large molecules that cannot cross by simple diffusion use specialized transmembrane carrier proteins that undergo conformational changes to shuttle the drug across.
- No energy required (no ATP)
- Can be saturated (limited number of carriers)
- Can be competitively inhibited by other substances using the same carrier
3. Active Transport
Carrier-mediated transport that works against the concentration gradient.
- Energy-dependent (ATP hydrolysis required)
- Saturable and competitively inhibited
- Selective for specific drug structures
- Example: absorption of levodopa via amino acid transporters in the gut
4. Endocytosis
Used for exceptionally large molecules that cannot otherwise cross the membrane.
- The cell membrane engulfs the drug, forming a vesicle that is pinched off into the cytoplasm
- Example: Vitamin B12 transported across the gut wall by endocytosis (via intrinsic factor receptors)
Factors Influencing Absorption
1. Effect of pH and Ionization (pH-Partition Hypothesis)
This is one of the most important determinants of GI absorption.
Most drugs are either weak acids or weak bases. A drug passes through lipid membranes more readily in its uncharged (nonionized) form.
- Weak acid (HA): HA ⇌ H⁺ + A⁻ — the nonionized form (HA) crosses membranes; the anion (A⁻) does not
- Weak base (BH⁺): BH⁺ ⇌ B + H⁺ — the uncharged free base (B) crosses membranes; the protonated form (BH⁺) does not
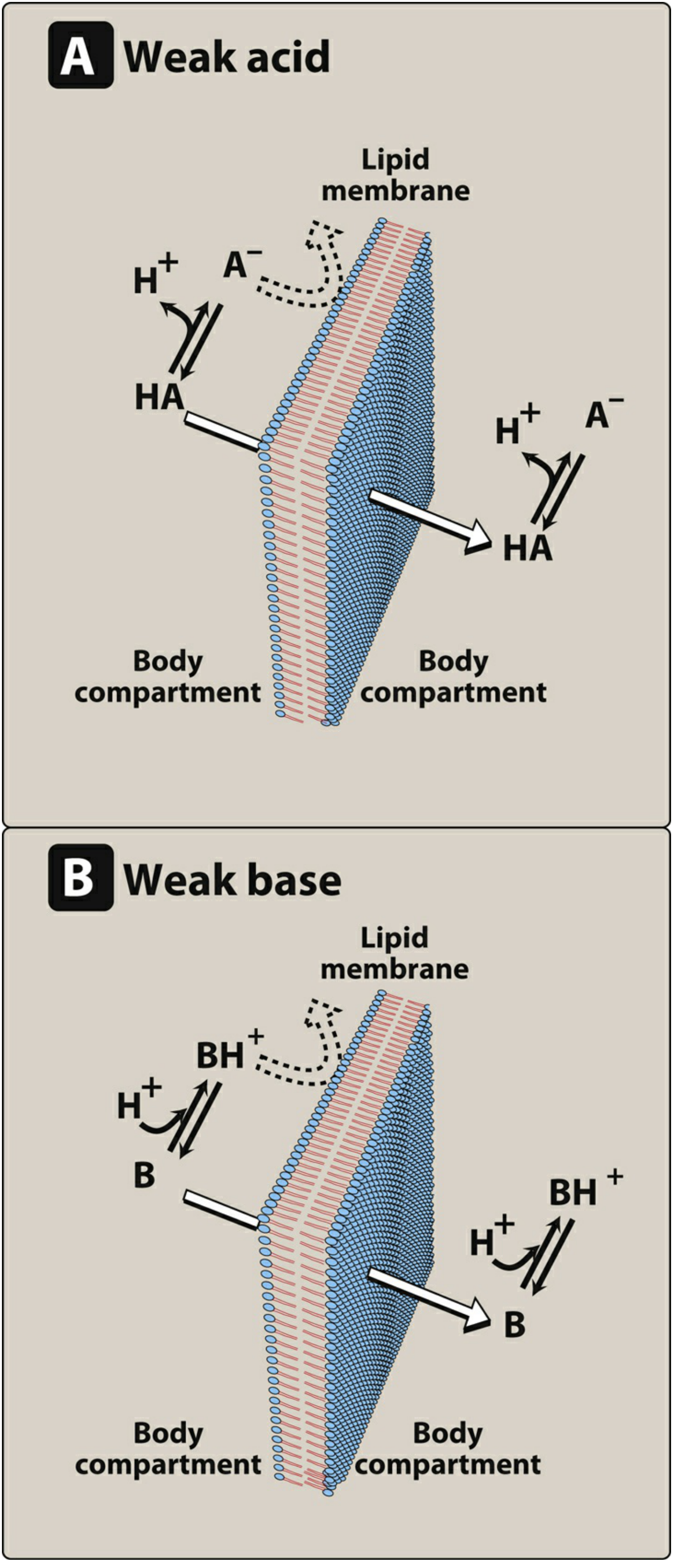
The ratio of ionized to nonionized forms at any given site is determined by the Henderson-Hasselbalch equation, which relates the ambient pH to the drug's pKa:
For weak acids: log([A⁻]/[HA]) = pH - pKa For weak bases: log([B]/[BH⁺]) = pH - pKa
Clinical implications:
- Weak acids (e.g., aspirin, pKa ~3.5) are predominantly nonionized in the acidic stomach (pH 1-2), so they are relatively well absorbed there - but the stomach's small surface area limits total absorption
- The upper intestine (pH 5-7) has a ~200 m² surface area, which overwhelmingly favors absorption of most drugs regardless of ionization
- Weak bases (e.g., morphine, pKa ~8) are more ionized in the stomach and better absorbed from the intestine
Ion trapping: When a drug is on one side of a membrane and the pH differs on the other side, the ionized (non-permeable) form accumulates on the more favorable side. For example, a basic drug taken orally will accumulate in the more acidic stomach lumen as BH⁺ - this is the principle behind ion trapping.
2. Blood Flow to the Absorption Site
The rate of absorption is proportional to local blood flow.
- The intestine receives far greater blood flow than the stomach, favoring intestinal absorption
- Shock severely reduces blood flow to cutaneous tissues - SC-administered drugs will be poorly absorbed in a shocked patient
- Highly perfused tissues (liver, kidney, brain, heart) receive drug fastest after systemic absorption
3. Total Surface Area Available for Absorption
- The small intestine, with its villi and microvilli (brush border), provides a surface area of approximately 200 m² - about 1000 times that of the stomach
- This enormous surface area makes the small intestine the primary site of oral drug absorption, even for drugs that are more ionized at intestinal pH
4. Contact Time at the Absorption Surface
- Decreased transit time (e.g., severe diarrhea, hypermotility) reduces absorption by shortening contact time
- Increased transit time (e.g., constipation) may increase absorption
- Gastric emptying rate controls delivery of drug to the intestine - delayed emptying (food, high-fat meals, anticholinergic drugs) slows absorption
- Food in the stomach both dilutes the drug and slows gastric emptying, generally slowing the rate (though not always the extent) of absorption
5. P-glycoprotein (Pgp) Efflux Pump
P-glycoprotein is a transmembrane ATP-dependent efflux transporter expressed in intestinal epithelial cells, hepatocytes, renal tubule cells, and the blood-brain barrier.
- It pumps drugs back out of cells into the intestinal lumen, reducing net absorption
- High P-gp expression in the gut wall significantly limits bioavailability of many drugs (e.g., digoxin, cyclosporine, certain antiretrovirals)
- P-gp is associated with multidrug resistance (MDR): cancer cells that overexpress P-gp pump out chemotherapeutic agents (paclitaxel, vinca alkaloids, anthracyclines), reducing their intracellular accumulation and causing treatment failure
6. First-Pass (Presystemic) Metabolism
After oral absorption, the drug enters the portal circulation and passes through the liver before reaching the systemic circulation. If the liver (or gut wall) rapidly metabolizes the drug during this initial passage, the amount of active drug reaching systemic circulation is substantially reduced.
Steps of presystemic elimination:
- Drug enters enterocyte → may be metabolized by CYP enzymes (CYP3A4) or effluxed back into gut lumen by P-gp
- Drug enters portal vein → transported to liver
- Liver metabolizes or excretes drug into bile → reduced systemic availability
Examples of high first-pass drugs:
| Drug | % Cleared First-Pass | Consequence |
|---|---|---|
| Nitroglycerin | >90% | Given SL, transdermal, or IV - not oral |
| Morphine | ~60-70% | Higher oral doses needed vs. IV |
| Lidocaine | ~70% | Not given orally |
| Propranolol | ~60-70% | Large oral doses; variable bioavailability |
7. Solubility of the Drug
For a drug to be absorbed, it must:
- Be lipid-soluble enough to cross lipid membranes
- Have some aqueous solubility to dissolve in GI fluids and reach the membrane surface
Both extremes are problematic:
- Highly hydrophilic drugs cannot cross the lipid bilayer
- Extremely lipophilic drugs are insoluble in aqueous fluids and cannot access the absorptive surface
This is why many drugs are formulated as weak acids or weak bases - they can exist in both ionized (aqueous-soluble) and nonionized (lipid-soluble) forms depending on the local pH.
8. Chemical Instability of the Drug
- Acid-labile drugs (e.g., penicillin G, omeprazole) are destroyed by gastric acid - require enteric coating or parenteral administration
- Enzyme-sensitive drugs (e.g., insulin, heparin, peptide drugs) are digested by proteases in the GI tract - cannot be given orally
9. Drug Formulation
Physical and pharmaceutical characteristics of the dosage form significantly affect absorption:
| Factor | Effect |
|---|---|
| Particle size | Smaller particles dissolve faster → faster absorption |
| Salt form | Different salts have different solubilities (e.g., chlorpromazine HCl vs. base) |
| Crystal polymorphism | Different crystal forms have different dissolution rates |
| Enteric coating | Delays dissolution until the intestine, protecting acid-labile drugs |
| Excipients | Binders, disintegrants, and dispersing agents alter dissolution rate |
| Extended-release formulations | Deliberately slow absorption to prolong drug action |
Bioavailability
Bioavailability (F) is the rate and extent to which an administered dose reaches the systemic circulation unchanged. It is determined by comparing the area under the plasma concentration-time curve (AUC) of the test route to that of IV administration (F = 1 by definition).
$$F = \frac{AUC_{oral}}{AUC_{IV}} = \frac{\text{Quantity reaching systemic circulation}}{\text{Quantity administered}}$$
Bioavailability is reduced by:
- Incomplete absorption from the GI tract
- First-pass hepatic and intestinal metabolism
- P-gp efflux
- Chemical instability
- Poor formulation dissolution
Bioequivalence: Two formulations are bioequivalent if they show comparable bioavailability (similar AUC) and similar times to peak plasma concentration (Tmax). Therapeutic equivalence additionally requires pharmaceutical equivalence (same active ingredient, dose, dosage form, and route).
Summary of Key Factors
| Factor | Direction of Effect |
|---|---|
| Nonionized (uncharged) form of drug | Increases absorption |
| Low pH for weak acids | Increases nonionized fraction → increases absorption |
| High pH for weak bases | Increases nonionized fraction → increases absorption |
| High blood flow at absorption site | Increases absorption |
| Large surface area (small intestine) | Increases absorption |
| Prolonged contact time | Increases absorption |
| P-glycoprotein expression | Decreases absorption |
| First-pass metabolism | Decreases bioavailability |
| Lipophilicity (moderate) | Increases absorption |
| Extreme hydrophilicity or lipophilicity | Decreases absorption |
| Food/delayed gastric emptying | Slows rate (may or may not affect extent) |
| Diarrhea/increased motility | Decreases absorption |
Sources:
- Lippincott Illustrated Reviews: Pharmacology, Sections III & IV (Absorption, Bioavailability)
- Harrison's Principles of Internal Medicine, Chapter 71 (Absorption and Bioavailability, First-Pass Effect)
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, Chapter 2 (Drug Absorption and Bioavailability)
This is a shared conversation. Sign in to Orris to start your own chat.