Explain the mechanism of urticaria
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Searching Images
urticaria mast cell degranulation IgE mechanism diagram
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compile a thorough answer.
Mechanism of Urticaria
Urticaria (hives) is fundamentally a disorder of cutaneous mast cell degranulation leading to transient dermal microvascular hyperpermeability, plasma extravasation, and sensory nerve activation — producing the classic wheal-and-flare response with pruritus. — Fitzpatrick's Dermatology, Robbins & Cotran Pathologic Basis of Disease
The Central Pathway: Mast Cell Degranulation
Dermal mast cells are the central effectors. When activated, they release:
Preformed (granule) mediators — released within minutes:
- Histamine — binds H1 receptors on postcapillary venule endothelium → vasodilation and increased vascular permeability → wheal; binds sensory nerves → pruritus
- Tryptase, chymase — serine proteases that amplify inflammation
Newly synthesized mediators — generated via arachidonic acid:
- Prostaglandins (PGD₂) — vasodilation, itch
- Leukotrienes (LTC₄, LTD₄) — potent vasodilators and bronchoconstrictors
- Platelet-activating factor (PAF) — increases vascular permeability
Cytokines (TNF-α, IL-4, IL-5, IL-13) — sustain the inflammatory response
The net result: vasodilation + plasma extravasation into the dermis = wheal (edema); surrounding erythema from arteriolar vasodilation (flare); pruritus from direct sensory nerve stimulation by histamine and kinins. — Rosen's Emergency Medicine
Classification by Mechanism
1. Mast Cell-Dependent, IgE-Dependent (Type I Hypersensitivity)
This is the classic allergic mechanism:
- Sensitization phase: On first antigen exposure (pollen, food, drugs, insect venom), Th2 cells produce IL-4 and IL-13, driving B-cell class switching → antigen-specific IgE is produced and binds to high-affinity FcεRI receptors on dermal mast cells and circulating basophils
- Elicitation phase: Re-exposure to the same antigen cross-links adjacent FcεRI-bound IgE molecules, triggering a signaling cascade:
- Activation of SYK → LAT → PLCγ → PKC
- Rise in intracellular Ca²⁺
- Explosive mast cell degranulation with release of histamine and other mediators
Symptoms typically develop within 15 minutes to 1 hour of antigen exposure. Despite being the best-understood mechanism, classical type I allergy accounts for less than 10% of all urticaria cases. — Fitzpatrick's Dermatology
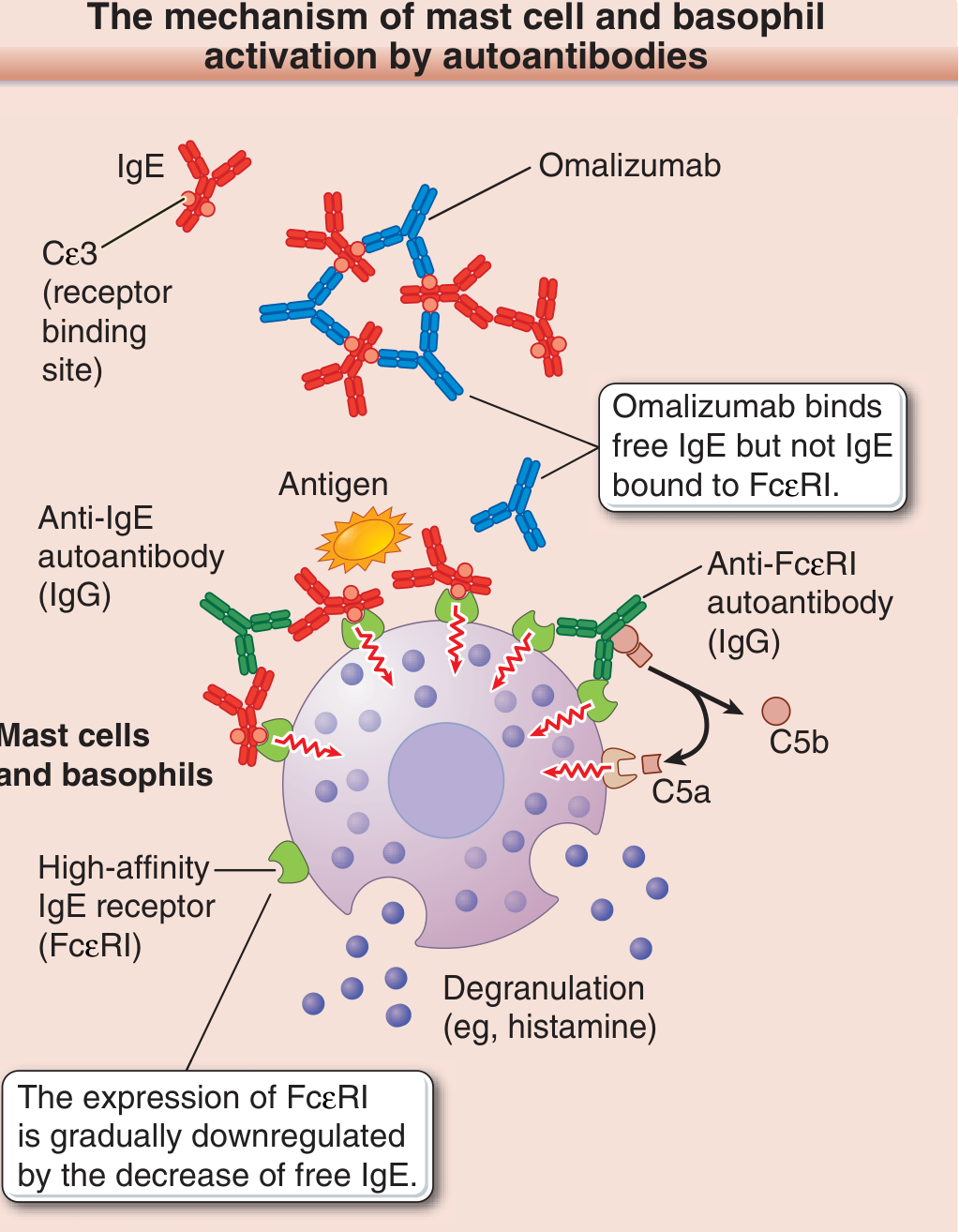
2. Autoimmune Mechanism (IgE-Dependent variant — Chronic Spontaneous Urticaria)
One-third to one-half of chronic spontaneous urticaria (CSU) cases have a detectable autoimmune basis:
- Anti-FcεRI IgG autoantibodies: Directly bind and cross-link the high-affinity IgE receptor on mast cells/basophils → degranulation (complement-dependent, via C5a)
- Anti-IgE IgG autoantibodies: Cross-link IgE itself while it is bound to FcεRI → same downstream result
- These can be detected by the autologous serum skin test (ASST) and the basophil histamine release test
- The complement fragment C5a amplifies degranulation
This explains why omalizumab (anti-IgE monoclonal antibody) works in CSU: by sequestering free IgE, it depletes IgE available to bind FcεRI, gradually downregulating FcεRI expression on mast cells, reducing their reactivity. — Fitzpatrick's Dermatology
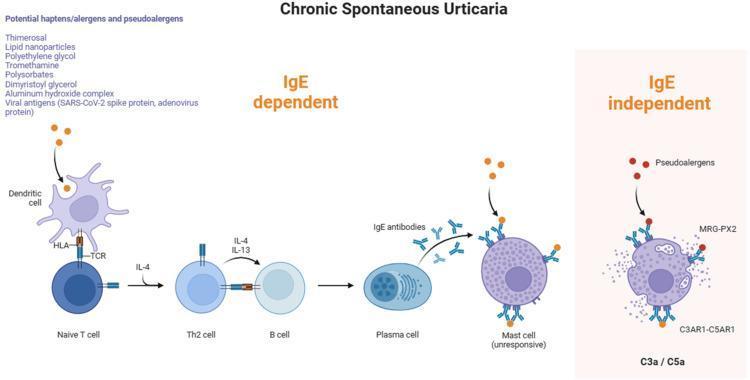
3. Mast Cell-Dependent, IgE-Independent
Certain substances directly trigger mast cell degranulation without prior IgE sensitization:
- Opiates (morphine, codeine), vancomycin, radiocontrast media — direct mast cell degranulation via MRGPRX2 receptor
- Foods (lobster, strawberries) — direct histamine release, non-immunologic
- No prior sensitization required; can occur on first exposure
4. Mast Cell-Independent, IgE-Independent
These mechanisms bypass mast cells altogether:
- Aspirin/NSAIDs: Inhibit cyclooxygenase (COX-1) → shunt arachidonic acid toward the lipoxygenase pathway → excess leukotrienes → vascular permeability. The precise mechanism is not fully elucidated, and effects can persist weeks after ingestion
- Hereditary angioedema (HAE): Deficiency of C1 inhibitor → unregulated activation of the early complement cascade and contact activation system → generation of bradykinin → binds B2 receptors on postcapillary venule endothelium → vasodilation and edema (notably mast cell-independent; antihistamines are ineffective in pure HAE)
- ACE inhibitors: Block bradykinin degradation → bradykinin accumulation → angioedema (same pathway as HAE)
— Robbins & Cotran Pathologic Basis of Disease
5. Neurogenic Mechanism
Cutaneous mast cells express receptors for numerous neuropeptides — substance P, vasoactive intestinal peptide (VIP), somatostatin, neurokinin A/B, CGRP, ACTH, and acetylcholine. In cholinergic urticaria, excessive acetylcholine release from cholinergic nerve terminals (in response to heat/exercise/sweating) directly activates mast cells. Two thirds of cholinergic urticaria patients are also sensitized to antigens in human sweat (specifically MGL_1304, a protein from Malassezia globosa), explaining the IgE-mediated overlap. — Fitzpatrick's Dermatology
Summary Table
| Mechanism | Key Mediator | Example Triggers | IgE? | Mast Cell? |
|---|---|---|---|---|
| Type I hypersensitivity | Histamine, leukotrienes | Foods, drugs, insect venom | ✅ Yes | ✅ Yes |
| Autoimmune (anti-FcεRI/anti-IgE IgG) | Histamine, C5a | Idiopathic CSU | ✅ Yes | ✅ Yes |
| Direct mast cell degranulation | Histamine | Opiates, contrast media | ❌ No | ✅ Yes |
| COX inhibition (aspirin) | Leukotrienes | NSAIDs | ❌ No | ✅ Yes |
| Bradykinin (HAE/ACEi) | Bradykinin | C1-inh deficiency, ACEi | ❌ No | ❌ No |
| Neurogenic | Histamine, acetylcholine | Heat, exercise, sweat | ± | ✅ Yes |
Histopathology
Regardless of mechanism, the histological features of urticaria are subtle:
- Widened collagen bundles (dermal edema)
- Dilated dermal lymphatics
- Sparse superficial perivenular infiltrate: mononuclear cells, rare neutrophils, ± eosinophils
- No epidermal changes (this distinguishes urticaria from eczema)
— Robbins & Cotran Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.