Neuromuscular junction, anatomy, physiology and pharmacological basis of anesthesia.
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have all the material needed for a comprehensive, well-illustrated answer. Let me compile it.
Neuromuscular Junction: Anatomy, Physiology, and Pharmacological Basis of Anesthesia
1. ANATOMY OF THE NEUROMUSCULAR JUNCTION
The neuromuscular junction (NMJ) is the specialized synapse between a motor nerve terminal and a skeletal muscle fiber. It has three structural components:
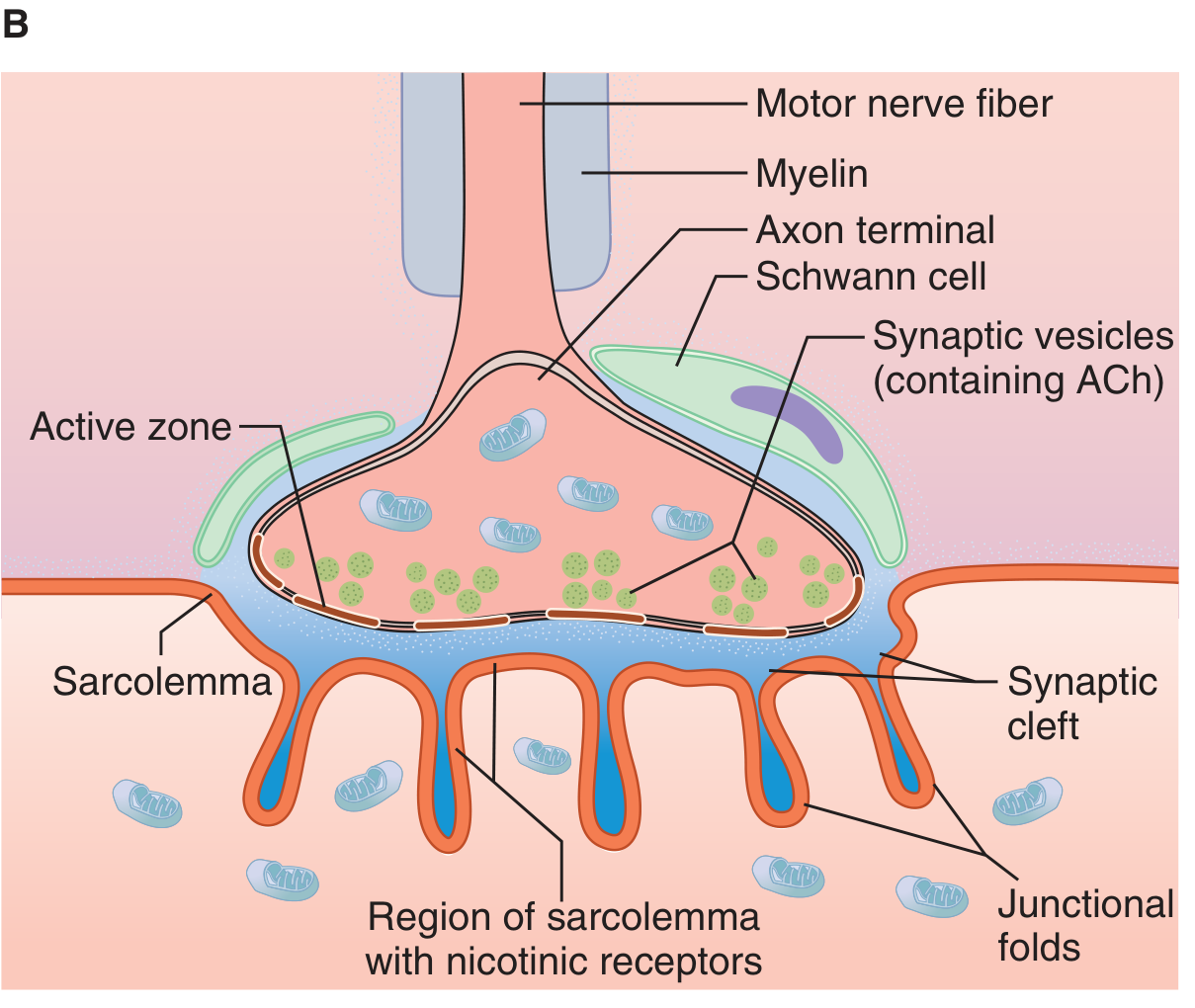
1.1 Presynaptic Component - Motor Nerve Terminal
- The motor axon loses its myelin sheath near its termination and divides into multiple terminal boutons
- The terminal contains mitochondria (for ATP-dependent ACh synthesis), and thousands of synaptic vesicles containing acetylcholine (ACh)
- Vesicle release occurs at specialized regions called active zones, which are rich in voltage-gated calcium channels (P-type channels) positioned immediately adjacent to the vesicle docking sites
- SNARE proteins (synaptobrevin/VAMP, syntaxin, SNAP-25) mediate vesicle docking and fusion
- Synaptotagmin serves as the calcium sensor on the vesicle membrane; synaptophysin is a glycoprotein component of the vesicle; synapsin phosphorylation facilitates vesicle trafficking to release sites
- Two pools of vesicles exist: a readily releasable pool (VP1) immediately adjacent to the active zone, and a reserve pool (VP2) farther away
(Miller's Anesthesia, 10e, p. 1160)
1.2 Synaptic Cleft
- Width approximately 50-70 nm
- Contains acetylcholinesterase (AChE), which rapidly hydrolyzes released ACh
- Basal lamina threads through the cleft
1.3 Postsynaptic Component - Motor Endplate
- The thickened sarcolemma at the junction, called the motor endplate, forms deep junctional folds
- The nicotinic ACh receptors (nAChR) are concentrated at the crests (tops) of these junctional folds at a density of up to 10,000/μm²
- AChE is anchored in the depths of the folds
- Each muscle fiber receives input from only a single motor nerve fiber (but one motor neuron can innervate many muscle fibers = motor unit)
2. PHYSIOLOGY OF NEUROMUSCULAR TRANSMISSION
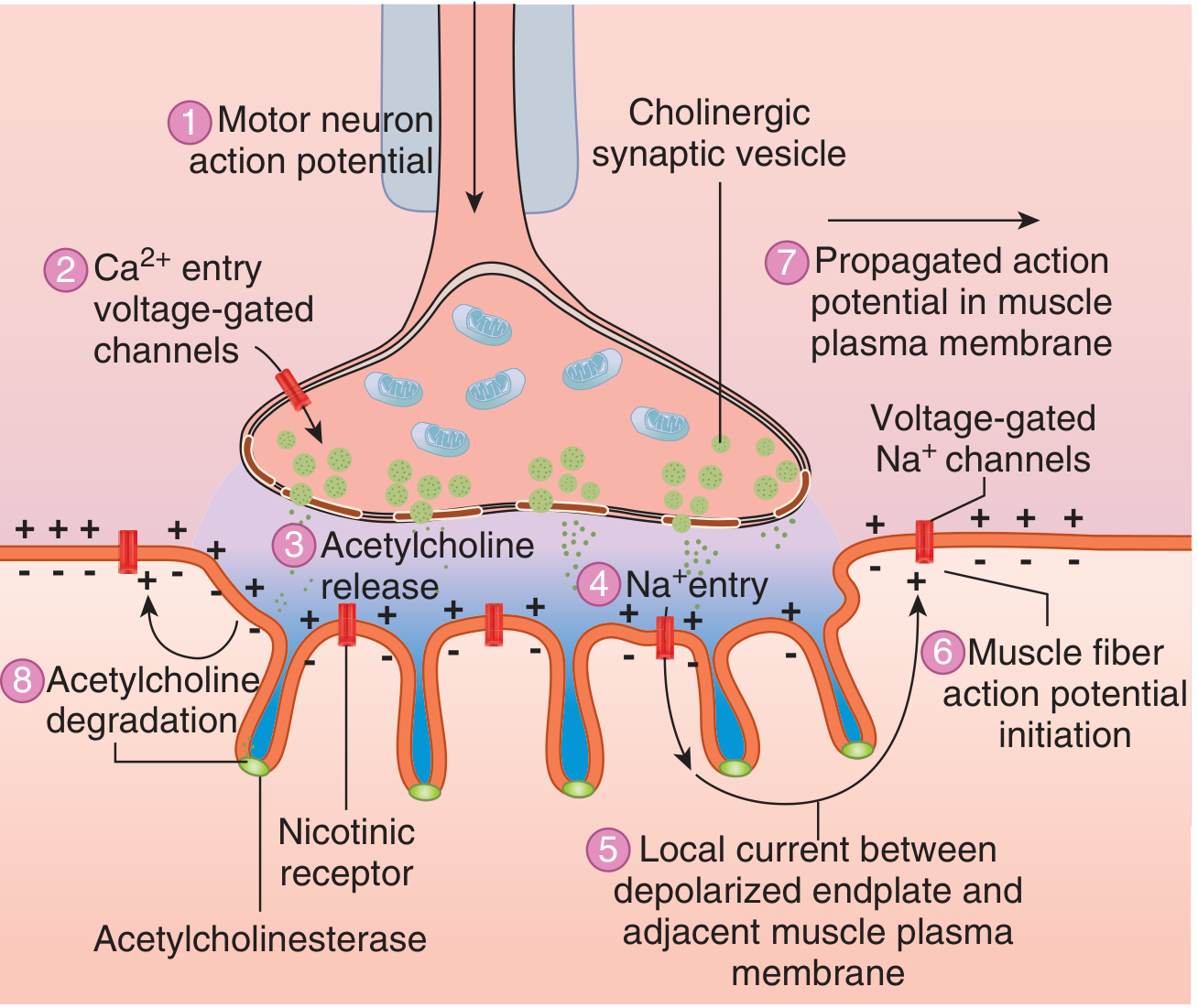
Step-by-Step Sequence of Transmission
Step 1 - ACh synthesis:
Choline (transported into the terminal via a sodium-coupled transporter) + acetyl-CoA (from mitochondria) → ACh, catalyzed by choline acetyltransferase (ChAT). ACh is packaged into vesicles (each vesicle = one quantum, ~10,000 molecules of ACh).
Step 2 - Action potential arrives:
A nerve action potential propagates down the axon to the terminal. Sodium influx depolarizes the membrane, opening voltage-gated calcium channels (P-type/Q-type) in the active zone.
Step 3 - Calcium entry triggers exocytosis:
Ca²⁺ enters the terminal and binds synaptotagmin. This activates the SNARE complex, causing vesicle fusion with the membrane and exocytosis of ACh quanta into the cleft. The number of quanta released is critically dependent on extracellular Ca²⁺ - doubling extracellular Ca²⁺ increases quantal content 16-fold. Mg²⁺ competes with Ca²⁺ and reduces transmitter release (explaining muscle weakness in patients receiving MgSO₄).
Step 4 - ACh diffuses and binds to nAChR:
ACh crosses the 50-70 nm cleft and binds to nicotinic M (NM) receptors at the tops of the junctional folds. Two molecules of ACh must bind simultaneously (to the two α-subunits) to open the ion channel.
Step 5 - End-plate potential (EPP) generated:
Channel opening allows Na⁺ influx and K⁺ efflux, producing a graded depolarization called the end-plate potential (EPP). If the EPP is sufficiently large, it depolarizes the adjacent muscle membrane above threshold.
Step 6 - Muscle action potential propagates:
A propagated action potential travels along the entire muscle fiber membrane (sarcolemma), leading to excitation-contraction coupling (Ca²⁺ release from sarcoplasmic reticulum → troponin-tropomyosin interaction → cross-bridge cycling → contraction).
Step 7 - ACh termination:
ACh is rapidly hydrolyzed by acetylcholinesterase (AChE) in the cleft into choline + acetate. Choline is recaptured by the nerve terminal for re-synthesis of ACh.
Safety Factor
Under normal conditions, the EPP is 3-4x larger than needed to fire an action potential. This large safety factor ensures reliable transmission. Neuromuscular blockers exploit this by reducing the EPP below the threshold for muscle excitation.
Posttetanic Potentiation (PTP)
During tetanic stimulation, Ca²⁺ accumulates in the nerve terminal (faster entry than clearance). This builds a Ca²⁺ surplus, causing a transiently supranormal ACh release for some time after the tetanus ends - this is the basis of PTP, clinically visible when a stimulated nerve in a partially paralyzed patient shows a bigger twitch after tetanic stimulation.
(Miller's Anesthesia, 10e, p. 1161)
3. THE NICOTINIC ACh RECEPTOR - MOLECULAR STRUCTURE
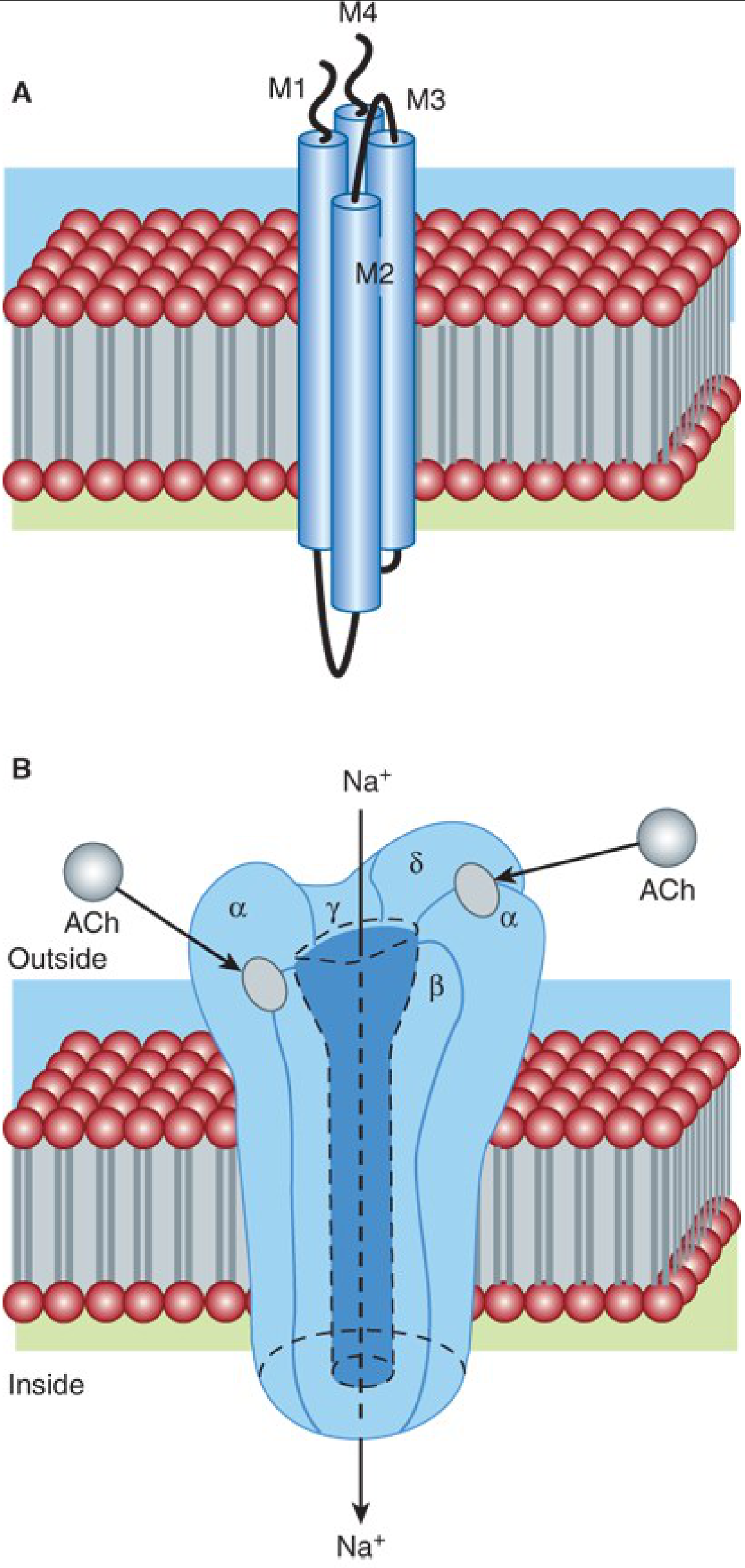
The nAChR is a pentameric ligand-gated ion channel with five subunits arranged around a central pore:
| Receptor Type | Subunit Composition | Location | Clinical Significance |
|---|---|---|---|
| Adult/Junctional | 2α₁, β₁, δ, ε | Endplate (innervated muscle) | Normal neurotransmission target |
| Immature/Extrajunctional (Fetal) | 2α₁, β₁, δ, γ | Extrajunctional (fetal, denervated, burns, immobilization, sepsis) | Opens longer, allows K⁺ efflux - causes hyperkalemia with succinylcholine |
| Neuronal α7 | 5α₇ (homopentamer) | Presynaptic nerve terminal | Modulates ACh mobilization/release |
- The M2 domain of each subunit lines the channel pore
- Two ACh molecules bind at the α-β and δ-α subunit interfaces to open the channel
- The channel is non-selective cation channel - primarily conducts Na⁺ inward (depolarizing) and K⁺ outward
(Katzung's Basic & Clinical Pharmacology, 16e, p. 743-744; Miller's Anesthesia, 10e, p. 1193-1194)
Extrajunctional receptor upregulation occurs after denervation, burns, prolonged immobilization, and sepsis. This dramatically increases the area of membrane responsive to depolarizing agents, explaining the life-threatening hyperkalemia with succinylcholine in such patients.
4. NEUROMUSCULAR BLOCKING DRUGS IN ANESTHESIA
NMBDs are used to:
- Facilitate tracheal intubation
- Provide optimal surgical relaxation
- Prevent patient movement during anesthesia
They are highly ionized compounds, inactive orally, and must be given parenterally. They do not cross the blood-brain barrier and therefore do not affect consciousness, cause sedation, or produce amnesia - a critical point because NMBD use has been implicated in accidental awareness under anesthesia.
4.1 Nondepolarizing Muscle Relaxants (NDMRs)
Mechanism: NDMRs competitively antagonize ACh at the α-subunits of the nAChR. A single molecule of NDMR occupying one α-subunit is sufficient to prevent channel opening (even if ACh binds the other site). As NDMR concentration rises, fewer channels open, the EPP diminishes, and neuromuscular block ensues.
Effect at Presynaptic Receptors: NDMRs also block presynaptic α3β2 neuronal nAChRs, which normally provide positive feedback to mobilize ACh from the reserve vesicle pool. Blocking these receptors reduces ACh mobilization, contributing to the characteristic "fade" seen with repetitive stimulation (train-of-four fade, tetanic fade).
Chemistry and Duration:
| Class | Drug | Duration | Elimination | Clinical Notes |
|---|---|---|---|---|
| Aminosteroid | Pancuronium | Long (>35 min) | Kidney (80%) | Vagolytic - tachycardia; avoid in renal failure |
| Vecuronium | Intermediate (20-35 min) | Liver (75-90%) | Minimal cardiovascular effects | |
| Rocuronium | Intermediate (20-35 min) | Liver (75-90%) | Fastest onset NDMR; choice for RSI if sugammadex available | |
| Pipecuronium | Long | Kidney | Less used now | |
| Benzylisoquinolinium | Atracurium | Intermediate (20-35 min) | Hoffmann elimination + ester hydrolysis | Organ-independent elimination; laudanosine metabolite |
| Cisatracurium | Intermediate (25-44 min) | Mostly spontaneous | Less histamine release than atracurium; ICU use | |
| Tubocurarine | Long (>50 min) | Kidney (40%) | Historical - histamine release, ganglion block | |
| Mivacurium | Short | Plasma cholinesterase | Short-acting NDMR; prolonged in cholinesterase deficiency |
(Katzung's Basic & Clinical Pharmacology, 16e, Table 27-1)
Pharmacokinetics: NDMRs have small volumes of distribution (80-140 mL/kg), roughly blood volume, as they are highly polar and do not readily cross cell membranes. Duration correlates with elimination half-life. Steroidal relaxants are hepatically metabolized to 3-hydroxy and 17-hydroxy metabolites (40-80% as potent) - these can accumulate with prolonged ICU use causing prolonged paralysis.
Potentiation of NDMRs by:
- Volatile anesthetic agents (halothane > enflurane > isoflurane > desflurane > sevoflurane)
- Aminoglycoside and polymyxin antibiotics (prejunctional and postjunctional effects)
- Hypermagnesemia (competes with Ca²⁺ at the nerve terminal)
- Hypothermia, respiratory alkalosis, hypokalemia
- Calcium channel blockers (modest potentiation of NDMR-induced block via L-type channels on nerve terminals)
4.2 Depolarizing Muscle Relaxants
Succinylcholine is the only clinically used depolarizing relaxant. Structurally, it is two molecules of ACh linked through their acetyl groups.
Phase I Block (Depolarizing Block)
Succinylcholine binds the nAChR and acts as an agonist - opening the channel and causing depolarization, visible as muscle fasciculations. Because succinylcholine is not hydrolyzed by AChE at the synapse (it is cleaved slowly by plasma cholinesterase/pseudocholinesterase), the membrane remains depolarized. Since excitation-contraction coupling requires repolarization ("repriming") of the endplate for repeated firing, a sustained depolarized, flaccid paralysis results. Single-channel recordings show succinylcholine causing prolonged "flickering" of the ion conductance at higher concentrations.
Features of Phase I Block:
- Fasciculations before paralysis
- No tetanic fade, no posttetanic facilitation
- Augmented (worsened) by cholinesterase inhibitors
- Onset <1 minute, duration 5-10 minutes
Phase II Block (Desensitization Block)
With repeated or prolonged succinylcholine exposure, the block transitions to a Phase II pattern resembling an NDMR block:
- Tetanic fade appears
- Posttetanic facilitation present
- Recovery takes >20 minutes
- The nAChR enters a desensitized state where it is unresponsive to agonists
- Reversal with cholinesterase inhibitors is unpredictable and not recommended
Comparison Table:
| Feature | NDMR (e.g., rocuronium) | Succinylcholine Phase I | Succinylcholine Phase II |
|---|---|---|---|
| Fasciculations | None | Yes | None |
| Tetanic response | Fade (unsustained) | Sustained | Fade (unsustained) |
| Posttetanic facilitation | Yes | No | Yes |
| Effect of neostigmine | Reversed | Augmented | Unpredictable |
| Recovery time | 30-60 min | 4-8 min | >20 min |
(Katzung's Basic & Clinical Pharmacology, 16e, Table 27-2)
Adverse Effects of Succinylcholine:
- Hyperkalemia: Normal rise 0.5-1.0 mEq/L. In burns, denervation (spinal cord injury), prolonged immobilization, crush injury - rise can exceed 5 mEq/L due to upregulated extrajunctional receptors. Risk begins 24-72 hours post-injury.
- Raised intraocular pressure (avoid in open-globe injury)
- Raised intragastric pressure (offset by raised lower esophageal sphincter tone)
- Raised intracranial pressure (controversial; often outweighed by benefit of faster intubation)
- Masseter spasm / Malignant hyperthermia (succinylcholine is a triggering agent in susceptible patients)
- Bradycardia (muscarinic effects, especially with repeated doses or in children)
- Prolonged block with pseudocholinesterase deficiency (dibucaine number testing identifies atypical enzyme)
5. MONITORING NEUROMUSCULAR BLOCK
Train-of-Four (TOF) Stimulation - four supramaximal stimuli at 2 Hz; count of twitches and TOF ratio assessed:
- TOF ratio <0.9 = clinically significant residual block (risk of aspiration, respiratory failure)
- TOF count 0 = profound block; 1-2 = deep block; 3-4 = moderate/shallow
Post-tetanic count (PTC) - used at deep block when TOF count = 0.
Residual paralysis (TOF ratio <0.9) occurs in approximately 40% of patients in the post-anesthesia care unit when not properly reversed, representing a significant clinical hazard.
(Barash Clinical Anesthesia, 9e, p. 1597)
6. REVERSAL OF NEUROMUSCULAR BLOCK
6.1 Acetylcholinesterase Inhibitors (Anticholinesterases)
Drugs: Neostigmine, pyridostigmine, edrophonium
Mechanism: Inhibit AChE → ACh accumulates in the cleft → ACh outcompetes NDMR for receptor binding (shifts the competitive equilibrium in favor of ACh). Neostigmine also has a minor direct effect of increasing ACh release from nerve terminals.
Critical limitation: Only effective at moderate block (TOF count ≥2). At deep block, the high NDMR concentration overwhelms the competitive reversal ability. Mathematically, doubling NDMR concentration requires a fourfold increase in ACh to remain competitive - hence anticholinesterases are ineffective at deep block.
Muscarinic side effects (bradycardia, bronchospasm, increased secretions) require co-administration of an anticholinergic (glycopyrrolate preferred with neostigmine; atropine with edrophonium).
Neostigmine weakness: Paradoxically, excess neostigmine can itself cause weakness by depolarizing block at the NMJ (too much ACh causes sustained depolarization). Maximum safe dose: 0.07 mg/kg.
6.2 Sugammadex - Selective Relaxant Binding Agent
Mechanism: A modified γ-cyclodextrin molecule with 16 inward-facing hydroxyl groups and 8 outward-facing carboxyl groups. It encapsulates rocuronium or vecuronium (not succinylcholine or benzylisoquinoliniums) in a tight 1:1 ratio. By binding free plasma drug, it creates a concentration gradient causing rocuronium/vecuronium to diffuse away from the NMJ back into the plasma, where it is immediately captured. The sugammadex-NMBD complex is excreted renally.
Key advantages:
- Works at any depth of block (unlike anticholinesterases, which fail at deep block)
- No muscarinic side effects - no need for anticholinergic co-administration
- Can reverse immediate post-intubation dose (16 mg/kg after 1.2 mg/kg rocuronium = "can't intubate, can't oxygenate" rescue strategy)
Dosing:
| Block Depth | Sugammadex Dose |
|---|---|
| Shallow (TOF count ≥2, T2) | 2 mg/kg |
| Deep (1-2 PTC, no TOF response) | 4 mg/kg |
| Immediate post-intubation (RSI rescue) | 16 mg/kg |
Adverse effects:
- Anaphylaxis (0.3% at 16 mg/kg dose)
- Bradycardia progressing to cardiac arrest
- Coagulopathy (~25% elevation of aPTT and PT/INR for up to 1 hour)
- Caution in severe renal impairment (CrCl <30 mL/min) - t½ extends to 19 hours; complex may not be eliminated effectively
(Katzung's Basic & Clinical Pharmacology, 16e, p. 759; Barash Clinical Anesthesia, 9e)
7. CLINICAL PHARMACOLOGICAL INTERACTIONS AT THE NMJ
| Drug/Condition | Effect at NMJ |
|---|---|
| Mg²⁺ (preeclampsia treatment) | Blocks P-type Ca²⁺ channels → ↓ ACh release → potentiates all NMBDs |
| Aminoglycosides | Pre- and postjunctional block; potentiate NDMRs |
| Volatile anesthetics | Enhance NDMR block (depth-dependent, agent-dependent) |
| Calcium channel blockers | Modest potentiation of NDMR block via L-type channels in nerve terminal |
| Eaton-Lambert syndrome | Autoantibodies against voltage-gated Ca²⁺ channels → ↓ ACh quantal release → sensitivity to both depolarizing and nondepolarizing NMBDs |
| Myasthenia gravis | Autoantibodies against nAChR → reduced receptor density → resistance to succinylcholine, extreme sensitivity to NDMRs |
| Burns/denervation | ↑ Extrajunctional receptors → hyperkalemia with succinylcholine; resistance to NDMRs |
Summary
The NMJ is a precisely organized chemical synapse where:
- ACh is synthesized, stored in quanta, and released in a Ca²⁺-dependent manner from the presynaptic terminal
- Released ACh binds pentameric nAChRs on the endplate, requiring two ACh molecules for channel opening, generating an EPP sufficient to fire a muscle action potential
- AChE terminates transmission within milliseconds
Anesthetic drugs exploit this:
- NDMRs (rocuronium, vecuronium, cisatracurium, etc.) competitively block nAChR, producing fade on repetitive stimulation
- Succinylcholine depolarizes the endplate but avoids hydrolysis at the synapse, producing sustained paralysis
- Reversal is achieved either by increasing ACh with anticholinesterases (neostigmine - limited at deep block) or by physically encapsulating steroidal NMBDs with sugammadex (effective at any block depth)
Sources: Miller's Anesthesia, 10e (pp. 1160-1194) | Ganong's Review of Medical Physiology, 26e (pp. 141-142) | Katzung's Basic & Clinical Pharmacology, 16e (pp. 743-759) | Barash's Clinical Anesthesia, 9e (pp. 1595-1597)
This is a shared conversation. Sign in to Orris to start your own chat.