Myocardial infraction
Reading File
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND "management"[MeSH Terms]
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
Reading File
I now have comprehensive data from multiple authoritative textbooks. Let me compile the full, detailed response.
Myocardial Infarction (MI)
Definition
Myocardial infarction (MI), commonly called a "heart attack," is death of cardiac muscle due to prolonged ischemia. The American College of Cardiology (ACC) and European Society of Cardiology (ESC) Fourth Universal Definition specifies that MI requires:
-
Acute myocardial injury (cardiac troponin rise/fall with at least one value above the 99th percentile upper reference limit), AND
-
At least one of:
- Symptoms of myocardial ischemia
- New ECG changes (ST/T wave changes or new Q waves)
- Imaging evidence of new loss of viable myocardium or wall motion abnormality
- Angiographic or autopsy evidence of coronary thrombus
-
Rosen's Emergency Medicine, 10th Ed.
Epidemiology
-
Approximately 800,000 MIs occur per year in the United States - nearly one every 40 seconds
-
Causes almost 400,000 deaths annually
-
10% occur in people under 40 years; 45% under 65 years
-
The major underlying cause is atherosclerosis
-
Male sex increases relative risk through middle age; females are generally protected during reproductive years due to estrogen; postmenopausal women see rapid acceleration of coronary artery disease (CAD) due to decreased estrogen, increased inflammatory markers, cholesterol, and blood pressure
-
Post-menopausal hormone replacement therapy has NOT been shown to be protective and may be detrimental (pro-thrombotic effect)
-
Robbins, Cotran & Kumar Pathologic Basis of Disease
Types of MI (Universal Classification)
| Type | Description |
|---|---|
| Type 1 | Spontaneous MI from plaque rupture/erosion/fissuring + thrombus (the "true ACS event") |
| Type 2 | MI secondary to supply-demand mismatch (spasm, embolism, severe anemia, arrhythmia, hypotension) |
| Type 3 | Sudden cardiac death before biomarker sampling; thrombus found at autopsy/angiography |
| Type 4 | MI associated with PCI (>3x 99th percentile URL defines it) |
| Type 5 | MI associated with CABG (>5x 99th percentile URL + new Q waves, new LBBB, or new occlusion) |
- Rosen's Emergency Medicine
Pathogenesis
Coronary Arterial Occlusion (Typical Sequence - ~90% of Cases)
- Atheromatous plaque erosion or rupture - exposes subendothelial collagen and necrotic plaque contents
- Platelet adhesion, aggregation, and activation - release of thromboxane A2, ADP, serotonin → further aggregation + vasospasm
- Coagulation cascade activation via tissue factor → growing thrombus
- Complete occlusion of the coronary artery lumen within minutes
- When angiography is performed within 4 hours of onset, coronary thrombosis is demonstrated in nearly 90% of cases. At 12-24 hours (without intervention), only 60% still show thrombosis (spontaneous lysis occurs in some)
Non-Atherosclerotic Causes (~10% of cases)
-
Vasospasm with or without atherosclerosis (e.g., cocaine, ephedrine)
-
Embolism - from left atrial thrombus (in AF), endocarditis vegetations, prosthetic material, or paradoxical embolism via patent foramen ovale
-
Uncommon causes: small vessel vasculitis, sickle cell disease, amyloid deposition, coronary artery dissection
-
Robbins, Cotran & Kumar Pathologic Basis of Disease
Cellular & Biochemical Response
The temporal progression of ischemic injury:
| Event | Timeframe |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | <2 minutes |
| ATP reduced to 50% normal | 10 minutes |
| ATP reduced to 10% normal | 40 minutes |
| Irreversible cell injury (necrosis) | 20-40 minutes |
| Microvascular injury | >1 hour |
| Full infarct extent | 6-12 hours |
- The first consequence is cessation of aerobic metabolism within seconds, leading to accumulation of lactic acid and depletion of creatine phosphate and ATP
- Only severe ischemia (flow ≤10% of normal) lasting 20-40 minutes leads to irreversible necrosis
- Irreversible necrosis first occurs in the subendocardial zone (most vulnerable - last to receive blood, exposed to highest intramural pressures), then spreads outward as a "wavefront" toward the epicardium
- Sarcolemmal disruption allows intracellular proteins (troponins, CK-MB) to leak into the circulation - the basis for biomarker testing
Cardiac muscle requires ~1.3 mL O₂/100 g/min just to survive; if even 15-30% of normal resting coronary flow is preserved, muscle will not die. The center of a large infarct with near-zero collateral flow dies.
- Robbins, Cotran & Kumar; Guyton & Hall Textbook of Medical Physiology
Patterns of Infarction
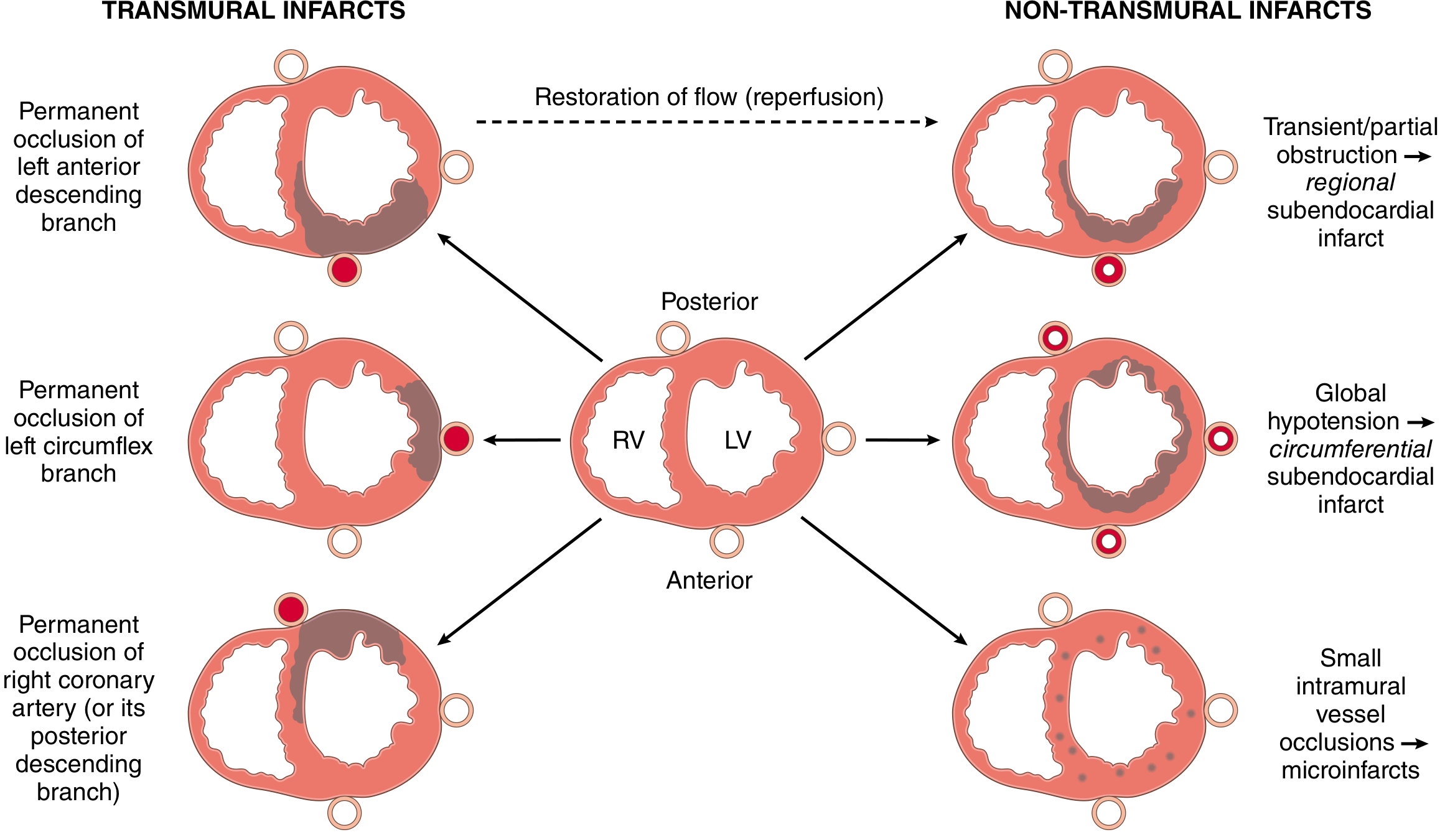
Fig. Distribution of MI patterns based on coronary artery involved (Robbins, Cotran & Kumar)
Coronary Artery - Territory Correlation
| Artery | Territory |
|---|---|
| LAD | Apex, anterior LV wall, anterior 2/3 of interventricular septum |
| RCA (in ~80% right-dominant circulation) | Entire RV free wall, posterobasal LV wall, posterior 1/3 of septum |
| LCX | Lateral wall of LV |
Infarct Patterns
- Transmural infarction - Full wall thickness necrosis; caused by complete, persistent occlusion of an epicardial vessel; associated with ST-elevation MI (STEMI)
- Subendocardial (nontransmural) infarction - Involves inner layers only; caused by transient/partial occlusion or plaque thrombus that lyses before full-thickness necrosis; also seen with global hypotension (circumferential subendocardial infarct); associated with NSTEMI
- Multifocal microinfarction - Pathology of small intramural vessels (microembolization, vasculitis, catecholamine excess)
Morphological (Gross & Microscopic) Changes
Gross Changes
| Time | Gross Appearance |
|---|---|
| 0-12 hours | Usually none visible; TTC staining reveals pallor/lack of staining |
| 12-24 hours | Pallor, possible slight yellowish discoloration |
| 1-3 days | Yellow-tan softening, pallor |
| 3-7 days | Hyperemic border; soft, yellow center with maximal pallor |
| 7-10 days | Depressed, soft; gelatinous reddish-gray at margins (granulation tissue) |
| Weeks | Grey-white fibrous scar forms at borders, progresses inward |
| >6-8 weeks | Dense white/grey fibrous scar |
Microscopic (Histological) Changes
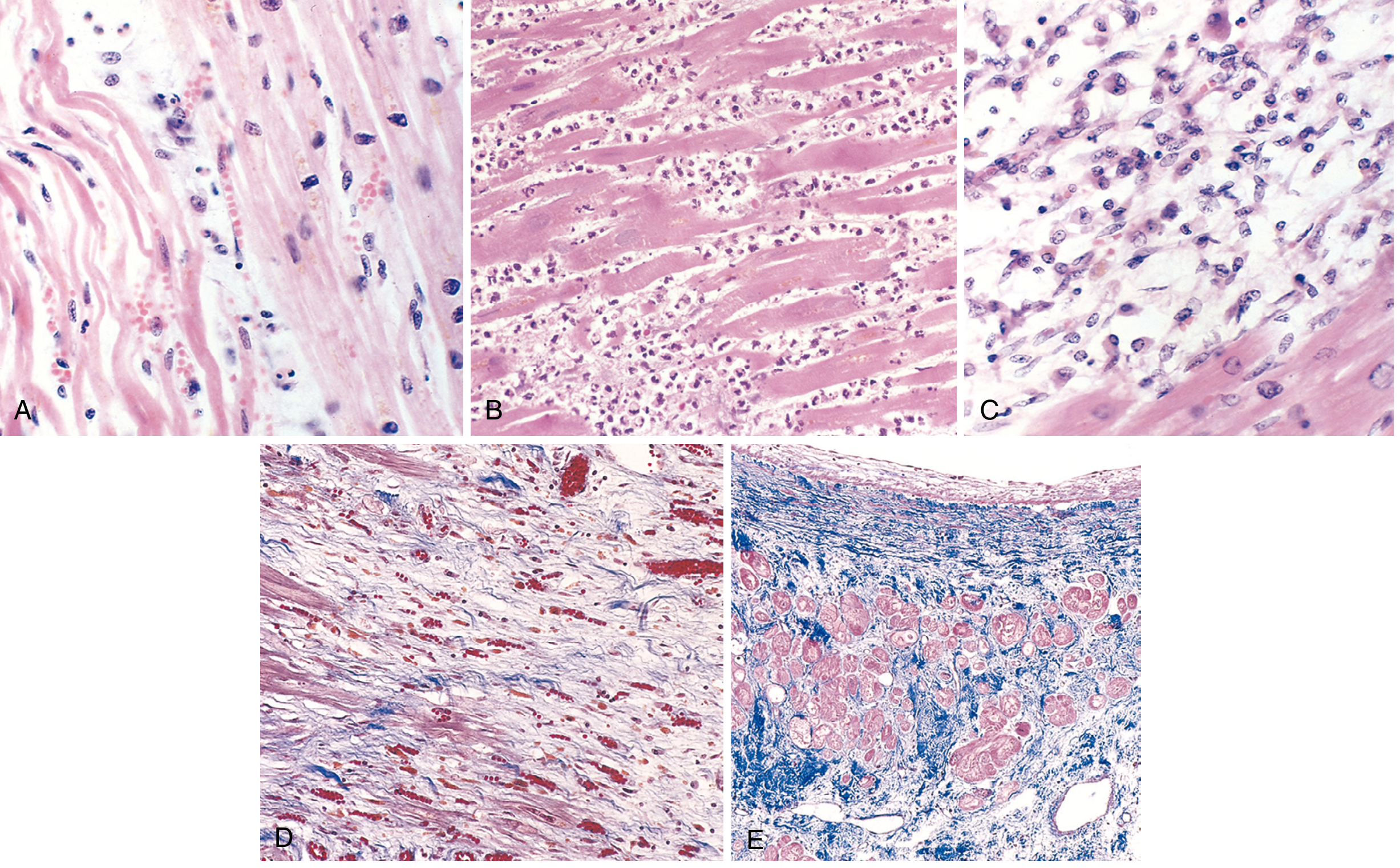
Fig. (A) Day 1: Coagulative necrosis with wavy fibers and scattered neutrophils. (B) Days 3-4: Dense neutrophilic infiltrate. (C) Days 7-10: Macrophage phagocytosis of necrotic debris. (D) Granulation tissue with loose collagen and new capillaries. (E) Healed infarct - dense collagenous scar with compensatory hypertrophy of residual muscle. (Robbins, Cotran & Kumar)
Key sequence:
- Coagulative necrosis with "wavy fibers" at infarct margins (Day 1)
- Neutrophilic infiltrate peaks at 1-3 days
- Macrophage cleanup peaks 3-7 days
- Granulation tissue forms 7-10 days (infarct heals from borders inward)
- Dense collagenous scar complete by ~6 weeks
- Note: Healed scars cannot be dated - an 8-week and a 10-year-old scar look identical
Reperfusion Injury
Restoration of flow after ischemia can cause additional injury:
- Contraction band necrosis - pathognomonic of reperfusion injury; irregular dense transverse bands of hypercontracted sarcomeres
- Gross appearance: Hemorrhagic infarct due to vascular leakage in the zone of reperfusion
- Despite this, reperfusion is still highly beneficial - the sooner achieved, the greater the myocardium salvaged
ECG Changes (Ganong's Physiology)
Three membrane abnormalities occur in acute MI:
| Defect in Infarcted Cells | Current Flow | ECG Change (leads over infarct) |
|---|---|---|
| Rapid repolarization (accelerated K⁺ channel opening) | Out of infarct | ST segment elevation |
| Decreased resting membrane potential (loss of intracellular K⁺) | Into infarct | TQ depression (recorded as ST elevation) |
| Delayed depolarization | Out of infarct | ST segment elevation |
Hallmark: ST segment elevation in leads overlying the infarct; ST depression in reciprocal leads.
- After days-weeks: ST abnormalities subside; dead muscle becomes electrically silent
- Dead area appears negative relative to normal myocardium during systole → pathologic Q waves develop (or R-wave regression in anterior leads)
- Non-Q-wave (NSTEMI) infarcts tend to be less severe but carry high risk of reinfarction
Clinical Presentation
Classic Symptoms
- Crushing, heavy, squeezing chest pain - typically substernal, radiating to left arm, jaw, neck, or back
- Pain lasting >20-30 minutes (distinguishes from stable angina)
- Diaphoresis, nausea, vomiting
- Dyspnea, anxiety, sense of impending doom
Atypical / Anginal Equivalent Presentations
-
One-third of ED patients with confirmed AMI have NO chest pain on presentation
-
Risk factors for atypical presentation: diabetes mellitus, older age (especially >85 years), female sex, nonwhite ethnicity, dementia, prior stroke/CHF
-
In patients >85 years, 60-70% present with anginal equivalents, especially dyspnea
-
Women: more likely to present with dyspnea, indigestion, weakness, unusual fatigue, cold sweats, anxiety
-
Diabetic patients: "silent MI" (medically unrecognized) occurs in ~40% vs. 25% in non-diabetics
-
Presentations that can mimic GERD, GI upset, or anxiety should prompt consideration of ACS
-
Rosen's Emergency Medicine
Biomarkers
| Marker | Rise | Peak | Return to Normal |
|---|---|---|---|
| Cardiac Troponin I/T (cTnI, cTnT) - preferred | 2-4 hours | 24-48 hours | 7-10 days |
| CK-MB | 3-8 hours | 18-24 hours | 48-72 hours |
| Myoglobin | 1-3 hours | 6-9 hours | 24 hours (least specific) |
-
Troponins begin to rise in 2-4 hours, peak at 24-48 hours, remain elevated 7-10 days
-
With reperfusion: troponin levels peak earlier and may be higher (rapid washout)
-
"Troponin leak" (low-level elevation) is seen in CHF, PE, renal failure, sepsis - serial measurements and clinical context help distinguish
-
Robbins, Cotran & Kumar
Causes of Death
- Decreased cardiac output - systolic stretch of non-contracting infarcted muscle further reduces effective pump function; in large infarcts (>40% of LV mass) → cardiogenic shock
- Pulmonary edema - LV failure → damming of blood in pulmonary vasculature
- Ventricular fibrillation - most common cause of sudden death early post-MI; due to:
- Abnormal action potentials at infarct border zone
- Re-entrant circuits from slowed conduction around infarcted area
- Ventricular dilation increasing conduction pathway length
- Cardiac rupture - occurs days 3-10 as necrotic muscle degenerates and thins; free wall rupture → cardiac tamponade → sudden death; septal rupture → VSD; papillary muscle rupture → acute mitral regurgitation
- Other arrhythmias: complete heart block (especially RCA occlusion affecting AV node), ventricular tachycardia
- Guyton & Hall Textbook of Medical Physiology
Complications
| Complication | Timing | Notes |
|---|---|---|
| Arrhythmias | Immediate - days | Most common cause of early death; VF, VT, complete heart block |
| Cardiogenic shock | Hours | LV dysfunction with low output |
| Papillary muscle dysfunction/rupture | Days 3-7 | Acute mitral regurgitation |
| Ventricular septal defect | Days 3-7 | RV ← LV shunt via septal rupture |
| Free wall rupture | Days 3-10 | Tamponade; usually fatal |
| LV thrombus | Days-weeks | Can embolize → stroke |
| Pericarditis (Dressler syndrome) | Weeks-months | Autoimmune post-MI pericarditis |
| Ventricular aneurysm | Weeks-months | Paradoxical motion on echo; persistent ST elevation |
| Heart failure (chronic) | Long-term | Remodeling, dilated cardiomyopathy |
Treatment
Initial (Acute) Management
- Oxygen - for hypoxia or respiratory distress (target SpO₂ ≥94%)
- Aspirin (antiplatelet) - immediately; inhibits TXA₂-mediated platelet aggregation
- P2Y12 inhibitor (clopidogrel, ticagrelor, or prasugrel) - dual antiplatelet therapy
- Anticoagulation - heparin (UFH or LMWH) or bivalirudin
- Nitroglycerin - for pain relief and preload reduction (avoid in RV infarct, hypotension, phosphodiesterase inhibitor use)
- Beta-blockers - reduce heart rate, oxygen demand; decrease risk of VF; start early if no contraindications (avoid in cardiogenic shock, severe bradycardia)
- Morphine - pain relief; used with caution (may mask symptoms, possible harm in NSTEMI)
Reperfusion (Primary Goal)
- STEMI: Primary percutaneous coronary intervention (PCI) is the gold standard; target door-to-balloon time <90 minutes
- If PCI not available within 120 minutes: thrombolysis (fibrinolytics: streptokinase, tPA, tenecteplase)
- Reperfusion is most beneficial when achieved quickly; benefits of early thrombolysis/angioplasty are well established
- NSTEMI/UA: Risk-stratify; early invasive strategy (angiography ± PCI) vs. conservative strategy based on TIMI/GRACE score
Long-term Management
-
ACE inhibitors (or ARBs) - reduce remodeling; especially in reduced EF
-
Beta-blockers - continued long-term; mortality benefit
-
Statins - high-intensity statin therapy regardless of baseline LDL
-
Dual antiplatelet therapy (DAPT) - for 12 months post-ACS/stent
-
Aldosterone antagonists (eplerenone, spironolactone) - in HFrEF post-MI
-
Cardiac rehabilitation - improves outcomes and quality of life
-
Robbins, Cotran & Kumar; Guyton & Hall; Rosen's Emergency Medicine
Recovery & Healing
-
After a large MI, dead fibers in the infarct center enlarge over days
-
Marginal collateral channels expand progressively, salvaging peri-infarct zone
-
Most nonfunctional muscle around the infarct eventually recovers over days to 3 weeks
-
In non-necrotic ischemic zones, pumping power typically returns to near-normal
-
The central necrotic area is replaced by fibrous scar tissue (complete by 6-8 weeks)
-
The remaining healthy muscle undergoes compensatory hypertrophy to maintain output
-
If >40% of LV mass is infarcted, adequate compensatory hypertrophy may be impossible, leading to progressive HF
-
Guyton & Hall Textbook of Medical Physiology
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease | Guyton & Hall Textbook of Medical Physiology | Rosen's Emergency Medicine | Ganong's Review of Medical Physiology | Robbins & Kumar Basic Pathology
This is a shared conversation. Sign in to Orris to start your own chat.