Explain all GIT disorders especially their Biochemistry
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Searching the Web
GIT gastrointestinal disorders biochemistry mechanisms 2024 review
I now have comprehensive material from the medical library. Let me compile the full, detailed response.
GIT Disorders: Comprehensive Biochemical Guide
1. PEPTIC ULCER DISEASE (PUD)
Normal Mucosal Defense Biochemistry
The gastric epithelium is protected by a multi-layered system:
- Mucus gel barrier: Secreted by mucous neck glands; forms a physical barrier between lumen and epithelium
- HCO3- secretion: Gastric epithelial cells secrete bicarbonate, which is trapped in mucus. Any H+ that penetrates is neutralized before reaching cells; pepsin is also inactivated in this alkaline microenvironment
- Prostaglandin E2 (PGE2): Maintains the mucosal barrier and stimulates HCO3- secretion - this is WHY NSAIDs cause ulcers (they inhibit COX enzymes, blocking prostaglandin synthesis)
- Mucosal blood flow and growth factors (EGF, TGF-alpha) round out protection
Pathogenesis: Biochemical Imbalance
PUD results from an imbalance between protective factors (mucus, HCO3-, PGE2, blood flow) and damaging factors (H+, pepsin, H. pylori, NSAIDs):
H. pylori Mechanisms:
- Contains urease enzyme: converts urea → NH3 + CO2. NH3 alkalinizes the local microenvironment, allowing bacterial survival in the acid lumen and promoting colonization
- NH4+ (in equilibrium with NH3) is directly cytotoxic
- Releases CagA toxin (cytotoxin-associated gene A), which disrupts epithelial tight junctions and breaks down the protective mucus barrier
- In the antrum: inhibits somatostatin secretion from D cells → removes inhibition of G cells → excess gastrin → excess H+ secretion (indirect route to duodenal ulcer)
- Spreads to duodenum → inhibits duodenal HCO3- secretion, removing the buffer against excess acid
- Diagnostic test: 13C-urea breath test - patient drinks 13C-labeled urea; H. pylori urease converts it to 13CO2, which is absorbed, expired, and measured
NSAID Mechanisms:
- Inhibit COX-1 (constitutively expressed) → reduced PGE2 synthesis → loss of mucosal protection, reduced HCO3-, reduced mucosal blood flow
- Also have direct topical irritant effects (weak acids that accumulate in epithelial cells)
Types & Biochemical Differences
| Feature | Gastric Ulcer | Duodenal Ulcer | Zollinger-Ellison |
|---|---|---|---|
| Primary defect | Defective mucosal barrier | Excess H+ secretion | Gastrin-secreting tumor |
| H+ secretion | Lower than normal (leaks back) | Higher than normal | Massively elevated |
| Gastrin | Elevated (due to reduced H+ feedback) | Elevated post-meal | Massively elevated (no feedback inhibition) |
| Parietal cell mass | Normal | Increased (trophic effect) | Markedly increased |
| H. pylori role | Direct - breaks barrier | Indirect - excess gastrin/reduced HCO3- | Not applicable |
Zollinger-Ellison Syndrome (Gastrinoma):
- Pancreatic islet cell adenoma secreting gastrin constitutively (not inhibited by luminal H+)
- Massive acid overload overwhelms duodenal HCO3- buffering
- Excess H+ inactivates pancreatic lipases → steatorrhea (fat maldigestion)
- Multiple ulcers from stomach through jejunum
- Treatment: H+ secretion inhibitors (omeprazole/PPIs - block H+/K+-ATPase; cimetidine - H2 receptor antagonist) + surgical tumor removal
Pepsinogen/Pepsin Biochemistry:
- Pepsinogen (inactive precursor) secreted by chief cells and mucous cells
- Activated to pepsin when gastric pH drops (autocatalytic below pH 5)
- Pepsin is a protease - contributes to mucosal self-digestion once the barrier is breached
- Pepsinogen secretion stimulated by: vagal stimulation (cephalic phase) + local H+ reflexes
(Robbins & Kumar Basic Pathology, pp. 569-571; Costanzo Physiology 7th Ed., pp. 370-374)
2. GASTRO-ESOPHAGEAL REFLUX DISEASE (GERD)
Biochemical Basis
- Lower esophageal sphincter (LES) incompetence allows gastric acid (H+, pH ~1-2) and pepsin to contact esophageal mucosa
- Esophageal epithelium lacks the mucous/HCO3- protective system of the stomach
- Repeated acid exposure:
- Activates transient receptor potential (TRP) channels in sensory neurons → heartburn
- Stimulates inflammatory cytokines (IL-8, IL-1β) → esophagitis
- Chronic exposure → metaplasia: Barrett's esophagus (columnar intestinal-type epithelium replaces squamous epithelium) - a pre-malignant change driven by aberrant gene expression (CDX2 transcription factor) and DNA methylation changes
- Peptic injury occurs in esophagus from acid reflux, or from ectopic gastric mucosa in a Meckel diverticulum
3. INFLAMMATORY BOWEL DISEASE (IBD)
IBD comprises two main conditions: Crohn Disease (CD) and Ulcerative Colitis (UC), both involving abnormal immune activation against intestinal microbiota.
Shared Biochemical Pathogenesis
Genetic Factors:
- NOD2 gene (Nucleotide Oligomerization Domain 2) - key susceptibility gene in Crohn disease
- NOD2 encodes a pattern recognition receptor that binds intracellular bacterial peptidoglycans and activates NF-kB
- Disease-associated NOD2 polymorphisms render it unable to effectively defend against intestinal bacteria → bacteria penetrate epithelium → trigger inflammatory cascades
- Autophagy-related genes (ATG16L1, IRGM): over 200 gene variants identified via GWAS; many encode components of the autophagosome pathway involved in handling intracellular bacteria
- Monozygotic twin concordance: ~50% in CD (strong genetic component) vs ~20% in UC (environmental factors more dominant)
Cytokine & Immune Imbalance:
- Trigger: microbial antigens presented to CD4+ Th cells → differentiation into Th1 and Th17 cells (driven by IL-12 and IL-23)
- Th1/Th17 cells activate macrophages, recruit neutrophils, release TNF-alpha, IFN-gamma, IL-17
- Crohn disease cytokine profile: IL-12, IL-23, IFN-gamma, TNF-alpha (Th1-dominated)
- Ulcerative colitis cytokine profile: Th2 shift with elevated IL-13 (causes epithelial damage)
- Regulatory T cell defects (especially IL-10-producing Tregs): rare mutations in IL-10 or IL-10 receptor → severe early-onset colitis
- Therapeutic targets: Anti-TNF antibodies (infliximab, adalimumab), anti-IL-12/23 antibodies (ustekinumab) - their efficacy confirms cytokine centrality
Epithelial Barrier Defects:
- Intestinal tight junction dysfunction in CD (cosegregates with NOD2 polymorphisms)
- Abnormal Paneth cell granules (contain antimicrobial peptides like defensins) → defective control of luminal microbiota composition
- Barrier dysfunction → antigen penetration → immune activation → more barrier damage (vicious cycle)
Microbiome:
- IBD patients show reduced microbial diversity and shifts in microbial composition (dysbiosis)
- Reduced protective bacteria (e.g., Faecalibacterium prausnitzii which produces anti-inflammatory butyrate)
- Increased proinflammatory bacteria → amplified mucosal immune responses
Crohn Disease - Specific Features
- Transmural inflammation (all layers involved)
- Skip lesions - discontinuous disease distribution
- Granuloma formation (noncaseating) in ~35% - driven by macrophage activation
- Terminal ileum most commonly affected → impairs B12 absorption (requires ileal intrinsic factor receptors) → megaloblastic anemia
- Fat malabsorption (bile salt recycling disrupted in terminal ileal disease) → steatorrhea, fat-soluble vitamin (A, D, E, K) deficiencies
- Risk of oxalate kidney stones: unabsorbed fatty acids bind calcium → free oxalate is absorbed (hyperoxaluria)
Ulcerative Colitis - Specific Features
- Mucosal/submucosal inflammation only; limited to colon
- Continuous disease from rectum proximally
- Risk of toxic megacolon (massive colonic dilation from transmural inflammatory spread)
- Elevated IL-13 causes epithelial apoptosis, reduced mucus production, and barrier breakdown
- Long-standing UC → dysplasia → colorectal cancer risk
(Robbins & Kumar Basic Pathology, pp. 576-584)
4. CELIAC DISEASE (Gluten-Sensitive Enteropathy)
Biochemical Pathogenesis
- Gluten (wheat protein) contains gliadin peptides that are resistant to full enzymatic digestion (proline-rich sequences resist intestinal proteases)
- Incompletely digested gliadin fragments cross the epithelial barrier (especially in genetically predisposed individuals with increased intestinal permeability)
- Tissue transglutaminase (tTG) in the lamina propria deaminates glutamine residues in gliadin peptides → modified peptides have high affinity for HLA-DQ2 and HLA-DQ8 molecules
- HLA-DQ2/DQ8 on antigen-presenting cells present deaminated gliadin to CD4+ Th1 cells → release of IFN-gamma, TNF-alpha → intestinal inflammation
- Autoantibodies: Anti-tTG IgA and anti-endomysial IgA (diagnostic markers); anti-gliadin antibodies also produced
- Anti-tTG antibodies cross-react with other tissues → extra-intestinal manifestations (dermatitis herpetiformis, cerebellar ataxia)
- Result: villous atrophy, crypt hyperplasia → massive reduction in absorptive surface area → global malabsorption
Consequences of Malabsorption (Biochemical)
| Malabsorbed substance | Deficiency consequence |
|---|---|
| Iron | Microcytic anemia |
| Folate | Megaloblastic anemia |
| Vitamin B12 | Megaloblastic anemia + neuropathy |
| Calcium + Vit D | Osteomalacia/rickets, secondary hyperparathyroidism |
| Vitamin K | Coagulopathy (PT prolonged) |
| Fats | Steatorrhea, fat-soluble vitamin deficiency |
| Proteins | Hypoalbuminemia, edema |
5. CARBOHYDRATE MALABSORPTION DISORDERS
Lactase Deficiency (Lactose Intolerance)
- Lactase (beta-galactosidase) at brush border cleaves lactose → glucose + galactose
- Congenital or acquired loss of lactase activity
- Unabsorbed lactose enters colon → bacterial fermentation → H2, CO2, short-chain fatty acids → osmotic diarrhea, bloating, cramping
- Breath hydrogen test (elevated H2 after lactose load) is diagnostic
Sucrase-Isomaltase Deficiency
- Autosomal recessive; sucrase-isomaltase complex on brush border is deficient
- Cannot cleave sucrose (glucose + fructose) or isomaltose (alpha 1-6 bonds from starch digestion)
- Results in osmotic diarrhea on sucrose ingestion
Glucose-Galactose Malabsorption
- Mutations in SGLT1 (Na+/glucose cotransporter) in brush border
- Cannot absorb glucose or galactose
- Presents at birth with severe osmotic diarrhea when fed glucose- or lactose-containing formulas
- Fructose (absorbed via GLUT5) is tolerated
6. PANCREATIC DISORDERS
Acute Pancreatitis
Biochemical Pathogenesis (Premature Enzyme Activation):
- Normally, digestive enzymes are synthesized as inactive zymogens (trypsinogen, chymotrypsinogen, proelastase, prophospholipase A2, prolipase) and activated only in duodenal lumen by enterokinase (cleaves trypsinogen → trypsin), which then activates the rest
- In pancreatitis, zymogens are prematurely activated within acinar cells:
- Trypsin activates other pro-enzymes → intracellular autodigestion cascade
- Phospholipase A2 activated → damages cell membranes, produces lysolecithin (toxic)
- Elastase activated → damages blood vessels → hemorrhage
- Lipase activated → fat necrosis (saponification - fatty acids combine with calcium → calcium soaps → hypocalcemia is a complication)
- Activated trypsin also stimulates complement, kinin, and coagulation cascades → systemic inflammatory response
Causes of Premature Activation:
- Gallstones blocking common bile duct (reflux of bile into pancreatic duct activates phospholipase A2)
- Alcohol (causes ductal obstruction, directly stimulates premature zymogen activation)
- Hypercalcemia (activates trypsinogen)
- Mutations in PRSS1 (cationic trypsinogen) - hereditary pancreatitis; trypsin becomes resistant to autolysis
Laboratory Changes:
- Serum amylase and lipase elevated (leak from damaged acinar cells)
- Hypocalcemia (calcium sequestered in fat necrosis)
- Elevated CRP, WBC (systemic inflammation)
Chronic Pancreatitis
- Repeated acute attacks or chronic alcohol → progressive fibrosis, loss of acinar cells, loss of islet cells
- Exocrine insufficiency: Reduced lipase, protease, amylase → steatorrhea, protein maldigestion, B12 malabsorption (R-binder proteins not degraded)
- Endocrine insufficiency: Loss of beta cells → secondary diabetes mellitus (pancreatogenic diabetes)
Cystic Fibrosis and GIT
- CFTR (Cl- channel) dysfunction → thick, viscous secretions in pancreatic ducts → obstruction → autodigestion
- GI manifestations: meconium ileus (newborns), exocrine pancreatic insufficiency, distal intestinal obstruction syndrome
7. MALABSORPTION SYNDROMES
Pathophysiological Classification
| Phase of Digestion | Disorder | Biochemical Defect |
|---|---|---|
| Luminal digestion | Chronic pancreatitis, Zollinger-Ellison | Reduced/inactivated enzymes |
| Mucosal absorption | Celiac disease, tropical sprue | Villous atrophy, reduced absorptive area |
| Lymphatic transport | Intestinal lymphangiectasia, Whipple's disease | Obstruction of chyle transport |
| Bacterial overgrowth | Blind loop syndrome | Bacteria deconjugate bile salts, compete for B12 |
Bile Salt Biochemistry in Malabsorption
- Bile salts are essential for micelle formation in the duodenum - solubilize dietary lipids and fat-soluble vitamins for absorption
- Primary bile acids (cholic acid, chenodeoxycholic acid) synthesized in liver from cholesterol (rate-limiting enzyme: cholesterol 7-alpha-hydroxylase)
- Conjugated with glycine or taurine → bile salts
- Reabsorbed in terminal ileum (enterohepatic circulation): 95% recycled, 5% lost in stool
- If terminal ileum is diseased/resected (Crohn disease): bile salt loss → reduced bile salt pool → fat malabsorption
Short Bowel Syndrome
- Massive small bowel resection → malabsorption of nearly everything
- Loss of brush border enzymes, transport proteins (SGLT1, GLUT2, CFTR, NHE3, PEPT1, etc.)
- Parenteral nutrition required until intestinal adaptation occurs (villous hypertrophy of remaining bowel driven by GLP-2)
8. DIARRHEAL DISORDERS
Osmotic Diarrhea
- Non-absorbable solutes in lumen → increased osmotic pressure → water drawn into lumen
- Examples: Lactase deficiency, Mg2+ laxatives, lactulose
- Stops with fasting
- High stool osmotic gap: Measured osmolality > 2x(Na + K)
Secretory Diarrhea
- Excess Cl- secretion or reduced Na+ absorption → obligatory water loss
- Cholera (Vibrio cholerae): CT-B subunit binds ganglioside GM1 on enterocytes; CT-A subunit ADP-ribosylates Gs-alpha → permanent activation of adenylyl cyclase → massive cAMP elevation → CFTR Cl- channels constitutively open → massive Cl- (and water) secretion → massive watery diarrhea (rice-water stools)
- E. coli LT toxin: Similar mechanism (ADP-ribosylates Gs)
- E. coli ST toxin: Activates guanylyl cyclase → elevated cGMP → activates CFTR
- VIPoma (Vasoactive Intestinal Peptide tumor): VIP activates adenylyl cyclase → watery diarrhea, hypokalemia, achlorhydria (WDHA syndrome)
- Continues despite fasting; normal osmotic gap
Invasive/Inflammatory Diarrhea
- Organisms (Salmonella, Shigella, C. difficile) invade or damage mucosa
- C. difficile: Toxin A (enterotoxin - disrupts tight junctions, induces apoptosis) + Toxin B (cytotoxin - inactivates Rho GTPases → cytoskeletal collapse → cell death) → pseudomembranous colitis
9. HEPATIC (LIVER) DISORDERS
Viral Hepatitis
- Hepatocyte injury biochemistry: Immune-mediated (CD8+ T cells kill infected hepatocytes) rather than direct viral cytopathicity (for HBV, HCV)
- Elevated ALT and AST (leak from damaged hepatocytes; ALT more liver-specific)
- Jaundice: impaired bilirubin conjugation and excretion
Bilirubin Metabolism (and its disruption):
| Step | Location | Enzyme | Disorder if blocked |
|---|---|---|---|
| Heme → biliverdin → unconjugated bilirubin | RES/spleen | Heme oxygenase | Hemolysis → unconjugated hyperbilirubinemia |
| Unconjugated bilirubin uptake | Hepatocytes | Ligandin (Y-protein) | Gilbert syndrome (UGT1A1 promoter mutation, reduced uptake + conjugation) |
| Conjugation: bilirubin + glucuronate | Hepatocyte ER | UGT1A1 (UDP-glucuronosyl transferase) | Crigler-Najjar type I (absent UGT1A1), type II (reduced), Gilbert syndrome |
| Excretion into bile canaliculi | Hepatocyte | MRP2 (canalicular transporter) | Dubin-Johnson syndrome (MRP2 mutation → conjugated bilirubin accumulates) |
| Intestinal conversion | Gut bacteria | Bacterial enzymes | - |
Cirrhosis
- End-stage of chronic liver injury: massive fibrosis, regenerative nodules
- Activated hepatic stellate cells (Ito cells) are the key fibrogenic cells - stimulated by TGF-beta1 to produce collagen type I and III → replacing functional parenchyma
- Portal hypertension (increased intrahepatic resistance due to fibrosis + active vasoconstriction by stellate cells)
- Biochemical consequences:
- Reduced albumin synthesis → hypoalbuminemia → ascites, edema
- Reduced clotting factor synthesis (II, V, VII, IX, X - all vitamin K-dependent factors, plus fibrinogen) → coagulopathy
- Reduced cholesterol metabolism, altered lipoprotein profiles
- Impaired drug metabolism (reduced CYP450 activity)
- Impaired ammonia detoxification (urea cycle): ammonia accumulates → hepatic encephalopathy (ammonia disrupts astrocyte function; glutamine synthesis in astrocytes causes astrocyte swelling/cerebral edema)
- Hyperaldosteronism (reduced aldosterone metabolism) → sodium and water retention
Non-Alcoholic Fatty Liver Disease (NAFLD/MASLD)
- Lipid accumulation in hepatocytes due to: insulin resistance → increased peripheral lipolysis (FFA flux to liver), increased de novo lipogenesis (elevated malonyl-CoA, SREBP-1c activation), impaired beta-oxidation and VLDL export
- Two-hit hypothesis: first hit (steatosis) + second hit (oxidative stress, mitochondrial dysfunction, inflammatory cytokines) → steatohepatitis → fibrosis
Wilson's Disease
- Autosomal recessive defect in ATP7B (copper-transporting ATPase in hepatocytes)
- Cannot incorporate copper into ceruloplasmin or excrete it into bile → copper accumulates in liver, brain, cornea (Kayser-Fleischer rings), kidneys
- Low serum ceruloplasmin, elevated urinary copper
Hemochromatosis
- HFE gene mutation (C282Y most common) → reduced hepcidin signaling → increased ferroportin activity on enterocytes → excess iron absorption
- Iron accumulates in liver (cirrhosis), pancreas (diabetes), heart (cardiomyopathy), skin (bronze pigmentation), gonads (hypogonadism) - "bronze diabetes"
- Elevated serum iron, transferrin saturation >45%, elevated ferritin
10. COLORECTAL CANCER (Biochemical Mechanisms)
Two main pathways:
- APC/WNT pathway (chromosomal instability): APC mutation → accumulation of beta-catenin → activation of WNT target genes (MYC, cyclin D1) → cell proliferation. Adenoma-carcinoma sequence (APC → KRAS → SMAD4/TGF-beta loss → TP53 mutation)
- Microsatellite instability pathway (MSI): Loss of DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2) → accumulation of mutations throughout genome → rapid carcinogenesis. Lynch syndrome is hereditary MMR deficiency.
11. ADDITIONAL NOTABLE GIT DISORDERS
Carcinoid Tumors (NETs)
- Arise from enterochromaffin cells; secrete serotonin (5-HT) from tryptophan via tryptophan hydroxylase + DOPA decarboxylase
- Carcinoid syndrome (when hepatic metastases present): flushing, diarrhea, bronchoconstriction from excess serotonin reaching systemic circulation
- Diagnosis: elevated urinary 5-HIAA (5-hydroxyindoleacetic acid, serotonin metabolite)
- Tryptophan diversion away from niacin synthesis → pellagra (niacin deficiency)
Whipple's Disease
- Infection with Tropheryma whipplei (gram-positive actinobactete)
- Infects macrophages in small bowel lamina propria; macrophages cannot digest bacteria (lysosomal defect)
- PAS-positive macrophages block lymphatics → lipid malabsorption, lymphedema
Hirschsprung's Disease
- Failure of neural crest cell migration → absence of ganglion cells (auerbach's myenteric and Meissner's submucosal plexuses) in distal colon
- Affected segment has tonic contraction (no inhibitory neurotransmission) → functional obstruction
- Loss of VIP and nitric oxide synthase (nNOS) neurons (inhibitory neurotransmitters) in affected segment
Ischemic Bowel Disease
- Bowel ischemia → reperfusion injury: xanthine oxidase generates reactive oxygen species (superoxide, H2O2, hydroxyl radical) on reperfusion → lipid peroxidation, DNA damage, cell death
- Mucosal injury releases bacterial endotoxin into portal circulation
Summary Biochemical Table
| Disorder | Key Biochemical Defect | Key Molecule/Enzyme |
|---|---|---|
| Peptic Ulcer | Imbalance H+/mucosal protection | H+/K+-ATPase, PGE2, mucus |
| Zollinger-Ellison | Ectopic gastrin → excess H+ | Gastrin, H+/K+-ATPase |
| H. pylori infection | Urease, CagA toxin | Urease, NH3, CagA |
| Celiac disease | HLA-DQ2/DQ8 + tTG-deaminated gliadin | Tissue transglutaminase, IL-15, IFN-gamma |
| Crohn disease | NOD2 mutations, Th1/Th17 cytokines | NOD2, NF-kB, IL-12, TNF-alpha |
| Ulcerative colitis | Th2 cytokines, IL-13 | IL-13, anti-epithelial autoimmunity |
| Cholera | ADP-ribosylation of Gs → cAMP↑ | Adenylyl cyclase, CFTR, cAMP |
| Acute pancreatitis | Premature zymogen activation | Trypsinogen, phospholipase A2, elastase |
| Lactase deficiency | Absent brush-border beta-galactosidase | Lactase |
| Crigler-Najjar | Absent UGT1A1 | UDP-glucuronosyltransferase |
| Gilbert syndrome | Reduced UGT1A1 (promoter mutation) | UGT1A1, bilirubin |
| Dubin-Johnson | Defective MRP2 transporter | MRP2, conjugated bilirubin |
| Wilson's disease | ATP7B mutation → copper accumulation | ATP7B, ceruloplasmin |
| Hemochromatosis | HFE mutation → excess iron absorption | HFE, hepcidin, ferroportin |
| Carcinoid | Excess 5-HT synthesis | Tryptophan hydroxylase, 5-HIAA |
| Colorectal Ca | APC/KRAS/TP53 or MMR loss | Beta-catenin, mismatch repair |
Sources:
- Robbins & Kumar Basic Pathology - Chapters 13 (GI pathology)
- Costanzo Physiology 7th Edition - Chapter 8 (GI physiology/biochemistry), pp. 369-376
- Goodman & Gilman's Pharmacological Basis of Therapeutics - GI pharmacology, IBD chapter
Explain bile disorders in full detail especially with reference to Biochemistry
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have comprehensive material from multiple authoritative textbooks. Let me compose the full detailed response.
Bile Disorders: Full Biochemical Guide
PART 1: NORMAL BILE BIOCHEMISTRY (Foundation)
1.1 Bile Acid Synthesis
Bile acids are synthesized exclusively in pericentral hepatocytes from cholesterol. This is the body's primary route for cholesterol elimination. The process involves 17 different enzymes organized into two major pathways:
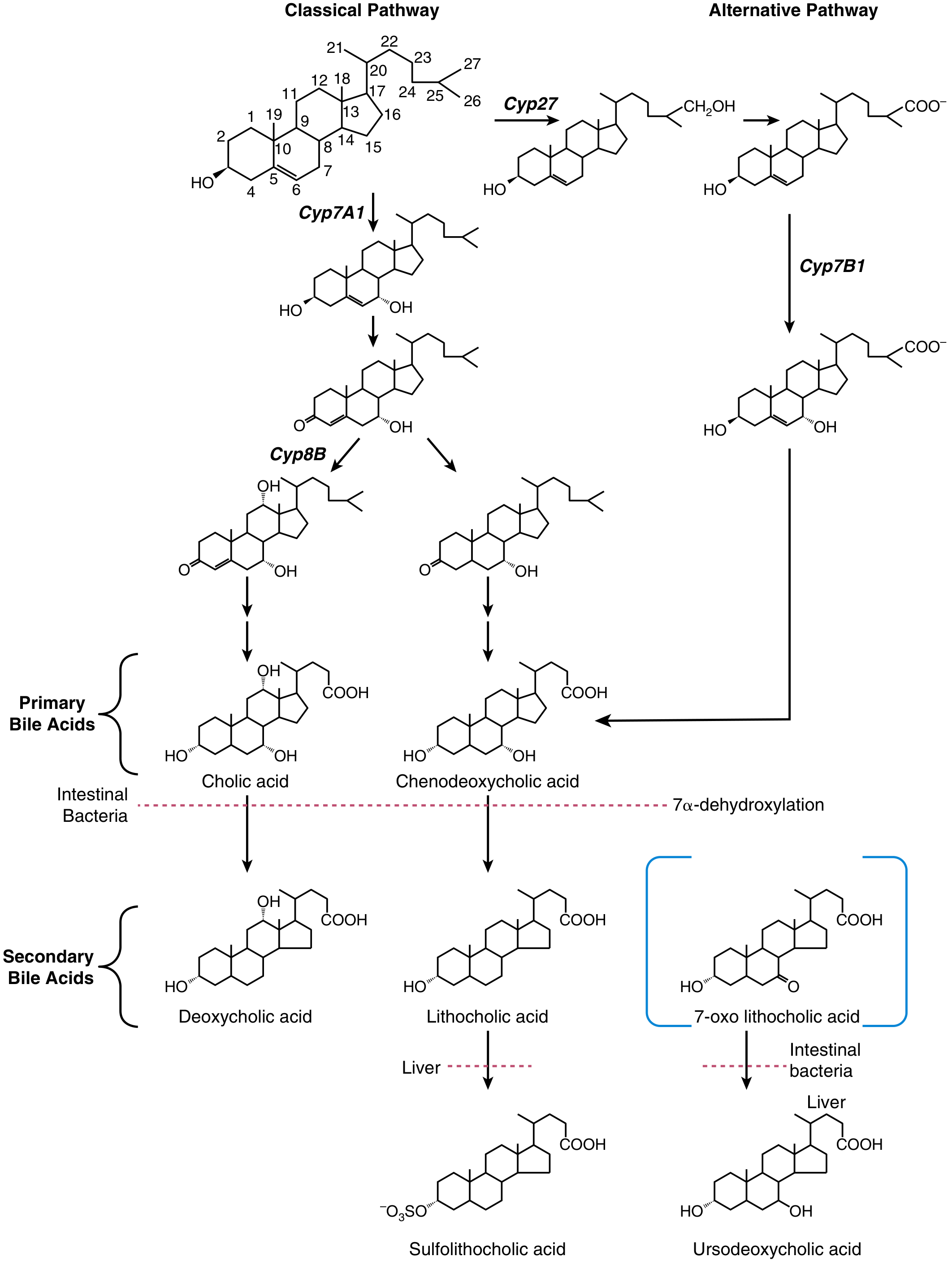
Classical (Neutral) Pathway - CYP7A1:
- Rate-limiting enzyme: cholesterol 7alpha-hydroxylase (CYP7A1) - hydroxylates cholesterol at C-7
- CYP8B1 (sterol 12alpha-hydroxylase) determines the ratio of cholic acid (CA) to chenodeoxycholic acid (CDCA)
- If CYP8B1 acts: produces cholic acid (CA) - trihydroxy bile acid (OH at C-3, C-7, C-12)
- If CYP8B1 is bypassed: produces chenodeoxycholic acid (CDCA) - dihydroxy bile acid (OH at C-3, C-7)
- Quantitatively dominant in adult humans (90% of bile acid production)
Alternative (Acidic) Pathway - CYP27/CYP7B1:
- Cholesterol is first hydroxylated on the side chain by CYP27A1 (sterol 27-hydroxylase) to form 27-hydroxycholesterol
- Then acted on by CYP7B1 (oxysterol 7alpha-hydroxylase)
- Favors CDCA biosynthesis
- Dominant in neonates (CYP7A1 is poorly expressed at birth)
- Mutations in CYP7B1 cause severe neonatal cholestatic liver disease
Normal synthesis rates: 0.2-0.6 g/day (inducible up to 4-6 g/day after small bowel resection)
1.2 Bile Acid Conjugation
Before secretion into the bile canaliculus, both CA and CDCA are N-acyl amidated (conjugated) with glycine or taurine:
- Enzyme: bile acid-CoA: amino acid N-acyltransferase (BAAT)
- First, bile acid is activated to a CoA thioester by bile acid-CoA ligase (BACL1/SLC27A5)
- Glycine conjugates predominate in humans (glycocholate, glycochenodeoxycholate)
- Taurine conjugates form a smaller fraction (taurocholate, taurochenodeoxycholate)
- Purpose of conjugation: Lowers pKa, keeps bile salts ionized and water-soluble at intestinal pH, increases amphiphilicity for micelle formation; also prevents passive reabsorption in the upper intestine
1.3 Secondary Bile Acid Formation (Gut Microbiota)
The intestinal microbiota transforms primary bile acids into secondary bile acids:
- 7alpha-dehydroxylation by colonic bacteria:
- Cholic acid → Deoxycholic acid (DCA)
- Chenodeoxycholic acid → Lithocholic acid (LCA) (most toxic; sulfated by liver for excretion)
- Bacterial bile salt hydrolases remove glycine/taurine from ~15% of bile acids in the small intestine (deconjugation)
- Ursodeoxycholic acid (UDCA) forms via epimerization of the C-7 hydroxyl of CDCA - the 7beta-isomer; it is hydrophilic and has cytoprotective/anti-apoptotic properties (used therapeutically)
1.4 Enterohepatic Circulation
The enterohepatic circulation is the most efficient recycling system in the body:
| Parameter | Value |
|---|---|
| Total bile acid pool | 2-4 g |
| Pool cycling per meal | 2-3 times |
| Total bile acid absorbed per day | 10-30 g |
| Fecal loss per day | 0.2-0.6 g |
| Efficiency of reabsorption | >95% |
Key transport proteins (from lumen to hepatocyte and back):
| Transporter | Gene | Location | Function |
|---|---|---|---|
| ASBT | SLC10A2 | Apical membrane of terminal ileal enterocytes | Active Na+-dependent uptake of conjugated bile acids from intestinal lumen |
| OST-alpha/beta | SLC51A/B | Basolateral membrane of enterocytes | Export bile acids into portal blood |
| NTCP | SLC10A1 | Sinusoidal membrane of hepatocytes | Na+-taurocholate cotransporting polypeptide; re-uptake of conjugated bile acids from portal blood (driven by Na+/K+-ATPase gradient) |
| OATP1B1/1B3 | SLCO1B1/3 | Sinusoidal membrane of hepatocytes | Na+-independent bile acid uptake (also takes up drugs, bilirubin) |
| BSEP | ABCB11 | Canalicular membrane of hepatocytes | Bile salt export pump; primary mechanism for bile acid secretion into bile canaliculus (ATP-dependent) |
| MRP2 | ABCC2 | Canalicular membrane of hepatocytes | Exports sulfated/glucuronidated bile acids, conjugated bilirubin, glutathione |
| MDR3 | ABCB4 | Canalicular membrane of hepatocytes | Flippase for phosphatidylcholine into bile (protects bile duct from detergent action of bile acids) |
| MDR1 | ABCB1 | Canalicular membrane | Exports drug metabolites |
| FIC1 | ATP8B1 | Canalicular membrane | P-type ATPase; maintains membrane integrity (aminophospholipid flippase) |
Micellar function in the intestine:
During a meal, intraluminal bile acid concentration reaches 5-10 mmol/L (above the critical micellar concentration of ~1.5 mmol/L). Bile acids form mixed micelles with phospholipids and cholesterol that solubilize dietary lipids (triglycerides, fat-soluble vitamins A, D, E, K) and their digestion products for absorption.
1.5 Feedback Regulation of CYP7A1 (FXR-FGF19 Axis)
This is one of the most elegant feedback systems in hepatology:
- After a meal, bile acids are absorbed by terminal ileum enterocytes
- Bile acids bind and activate FXR (Farnesoid X Receptor) - a nuclear receptor
- FXR in ileal enterocytes induces synthesis of FGF19 (fibroblast growth factor 19)
- FGF19 is secreted into portal circulation, reaches liver, and binds FGFR4/beta-klotho receptor complex on hepatocytes
- FGFR4 signaling represses CYP7A1 transcription - inhibiting further bile acid synthesis
- Additionally, FXR in hepatocytes induces SHP (small heterodimer partner) - an orphan nuclear receptor that inhibits HNF4alpha and LRH-1 (both are positive regulators of CYP7A1)
- FXR also induces ZFP36L1 (RNA-binding protein) → promotes CYP7A1 mRNA degradation
This dual (intestinal + hepatic) feedback ensures bile acid homeostasis. Disruption of this axis underlies multiple bile disorders.
PART 2: DISORDERS OF BILIRUBIN METABOLISM
Bilirubin is a breakdown product of heme - understanding its metabolism is essential for all bile disorders involving jaundice.
Complete Bilirubin Pathway
Hemoglobin (heme)
↓ [Heme oxygenase - RES/macrophages, spleen]
Biliverdin (green)
↓ [Biliverdin reductase]
Unconjugated bilirubin (UCB) - lipid-soluble, tightly bound to albumin
↓ [Uptake into hepatocyte via OATP; bound to ligandin/Y-protein]
Unconjugated bilirubin (free, in hepatocyte)
↓ [UGT1A1 - UDP-glucuronosyltransferase in hepatocyte ER]
Conjugated bilirubin (bilirubin diglucuronide) - water-soluble
↓ [MRP2 - canalicular excretion into bile]
Bile → Intestine
↓ [Bacterial enzymes in colon]
Urobilinogen (some reabsorbed, appears in urine; most oxidized to urobilin → stercobilin → brown stool color)
2.1 Gilbert Syndrome
- Prevalence: 5-10% of population (most common inherited disorder of bilirubin metabolism)
- Defect: Mutation in the UGT1A1 gene promoter (TA repeat in the TATA box: (TA)7TAA instead of normal (TA)6TAA) → reduced transcription → ~30% of normal UGT1A1 activity
- Also: mildly reduced hepatic uptake of bilirubin
- Result: Mild unconjugated hyperbilirubinemia (usually <3 mg/dL), exacerbated by fasting, stress, illness, exercise
- Benign: No liver damage, no treatment needed
- Important pharmacological note: UGT1A1 also conjugates drugs (e.g., irinotecan, statins) - Gilbert patients have reduced drug glucuronidation → drug toxicity risk (e.g., irinotecan-induced diarrhea)
2.2 Crigler-Najjar Syndrome
Complete or near-complete absence of UGT1A1:
| Feature | Type I | Type II |
|---|---|---|
| UGT1A1 activity | Absent (0%) | Residual (<10%) |
| Bilirubin level | 20-45 mg/dL | 6-20 mg/dL |
| Inheritance | AR | AR or AD |
| Jaundice | Severe, neonatal | Less severe |
| Kernicterus | Inevitable without treatment | Rare |
| Treatment | Phototherapy 12-16 h/day; liver transplant | Phenobarbital (induces UGT1A1) |
| Gene mutation | Various UGT1A1 coding mutations | Usually promoter/missense |
- Kernicterus mechanism: UCB crosses blood-brain barrier (lipid-soluble when unbound from albumin) → deposits in basal ganglia → uncouples oxidative phosphorylation → neuronal death
2.3 Dubin-Johnson Syndrome
- Defect: Loss-of-function mutations in MRP2 (ABCC2) - the canalicular multispecific organic anion transporter
- Result: Conjugated bilirubin cannot be exported into bile canaliculus → refluxes back into blood → conjugated (direct) hyperbilirubinemia
- Mild jaundice; benign prognosis
- Characteristic: Black liver (lysosomal accumulation of dark pigment - a polymer of epinephrine metabolites that cannot be exported)
- Urine coproporphyrin pattern: total normal, but >80% is coproporphyrin I (normally majority is III)
- BSP (bromsulphthalein) dye test: biphasic curve with secondary rise at 90 min (reflux of conjugated BSP back into plasma)
2.4 Rotor Syndrome
- Defect: Mutations in OATP1B1 and OATP1B3 (sinusoidal uptake transporters) → impaired hepatic reuptake of conjugated bilirubin that has refluxed into plasma
- Result: Conjugated hyperbilirubinemia
- Liver appears normal (no black pigment - unlike Dubin-Johnson)
- Urine coproporphyrin: total elevated, normal ratio (I:III approximately equal)
- BSP: absent secondary rise (no reflux mechanism)
2.5 Neonatal Physiologic Jaundice
- Unconjugated hyperbilirubinemia in newborns (days 2-7)
- Mechanisms:
- Accelerated hemolysis (fetal Hb replacement with adult Hb)
- Immature UGT1A1 activity (also reduced UDP-glucuronate synthesis)
- Reduced ligandin (Y-protein) levels
- Increased enterohepatic circulation of bilirubin (intestinal beta-glucuronidase deconjugates bilirubin → reabsorbed)
- Treatment: Phototherapy (converts UCB to water-soluble photoisomers excreted in bile/urine without conjugation)
PART 3: GALLSTONE DISEASE (CHOLELITHIASIS)
Gallstones form through three interacting mechanisms: bile supersaturation, crystal nucleation, and gallbladder dysmotility.
3.1 Cholesterol Gallstones (75-80% of stones in Western countries)
Bile is a unique thermodynamic system:
- Cholesterol is water-insoluble; it is solubilized in bile by mixed micelles (bile acids + phospholipids/lecithin) and vesicles (phospholipid bilayer particles)
- The cholesterol saturation index (CSI) = (actual cholesterol) / (maximum solubilizable cholesterol)
- CSI >1 = supersaturated bile = lithogenic bile
Step 1 - Bile Supersaturation:
- Excess cholesterol secretion (obesity, metabolic syndrome, estrogens)
- Estrogens increase hepatic LDL receptor expression → increased cholesterol uptake → increased biliary cholesterol secretion; also increase HMG-CoA reductase activity
- Oral contraceptives and pregnancy increase risk significantly
- Reduced bile acid pool (ileal disease/resection reduces enterohepatic cycling → less CDCA/CA → less micellar solubilization)
- Reduced phospholipid secretion (MDR3/ABCB4 mutations)
Step 2 - Crystal Nucleation:
- Supersaturation alone is insufficient - crystal nucleation must occur within the mucin gel layer
- Pronucleating factors: mucin, certain immunoglobulins, pigment particles, calcium ions
- Antinucleating factors: Apolipoprotein A-I, Apolipoprotein A-II, certain glycoproteins
- UGT1A1 Gilbert variant is associated with gallstone disease - pigment particles (from incomplete bilirubin conjugation) act as nucleating foci
- Vesicle fusion → liquid crystals → solid cholesterol monohydrate crystals → crystal growth
Step 3 - Gallbladder Dysmotility:
- Impaired gallbladder emptying allows crystals to remain and grow into macroscopic stones
- CCK-mediated contraction is reduced in lithogenic states
- FGF19-TGR5 axis normally induces gallbladder filling/relaxation during fasting; impaired FGF19 signaling contributes
- High-risk states: pregnancy (progesterone reduces smooth muscle contraction), total parenteral nutrition (no enteral fat → no CCK release → gallbladder stasis), rapid weight loss, spinal cord injury
Genetics:
- ~25% genetic contribution
- ABCG5/ABCG8 variants → increased biliary cholesterol secretion
- CYP7A1 variants → reduced bile acid synthesis
Stone types:
- Pure cholesterol stones: pale yellow, radiolucent, ovoid, often single
- Mixed stones: more common, contain calcium carbonate/phosphates/bilirubin (some radiopaque)
3.2 Pigment Gallstones (20-25%)
Form when unconjugated bilirubin concentration in bile exceeds its solubility:
Black pigment stones:
- Sterile gallbladder bile
- Made of calcium bilirubinate polymer + calcium phosphate + calcium carbonate
- Mechanisms:
- Chronic hemolysis (sickle cell disease, thalassemia, hereditary spherocytosis) → excess UCB production exceeds liver's conjugating capacity → elevated UCB in bile
- Cirrhosis → reduced bile acid synthesis → reduced solubilization of bilirubin → precipitation
- Small, numerous, very hard, brittle
- Radiodense (calcium-containing)
Brown pigment stones:
- Found in infected bile ducts (intrahepatic or extrahepatic)
- Bacteria (E. coli, Bacteroides, Clostridium) produce beta-glucuronidase → deconjugates conjugated bilirubin in bile → forms insoluble calcium bilirubinate
- Soft, mushy, earthy-brown
- Associated with: bile duct strictures, choledochal cysts, Caroli disease, biliary parasites (Clonorchis sinensis, Ascaris)
3.3 Biliary Sludge
- Precursor to gallstones; contains lecithin-cholesterol liquid crystals, cholesterol monohydrate crystals, calcium bilirubinate, mucin gel
- Implies: (1) deranged mucin secretion/elimination balance; (2) nucleation of biliary solutes
- In 14% of patients, progresses to frank gallstones
3.4 Acute Cholecystitis
- Usually triggered by a stone obstructing the cystic duct
- Mechanical obstruction → bile stasis → increased intraluminal pressure → mucosal ischemia
- Biochemical cascade:
- Concentrated bile (lithocholic acid, deoxycholic acid) causes mucosal damage (detergent action on cell membranes)
- Phospholipase A2 in bile cleaves lecithin → lysolecithin (directly toxic to mucosa)
- Prostaglandins (especially PGE2, PGI2) accumulate → inflammation, pain, fever
- Secondary bacterial invasion (E. coli, Klebsiella, Enterococcus) in ~50% of cases
3.5 Choledocholithiasis and Ascending Cholangitis
- Stone in common bile duct → obstructive jaundice
- Charcot's triad: RUQ pain + fever + jaundice
- Cholestasis → alkaline phosphatase (ALP) and gamma-glutamyl transpeptidase (GGT) most prominently elevated (from cholestatic injury to cholangiocytes)
- Conjugated bilirubin elevated (secreted into bile but cannot pass obstruction → refluxes into blood via MRP3 on hepatocyte sinusoidal membrane)
- Bile acid accumulation in plasma → pruritus (bile acids deposit in skin, activating TGR5 and TRPV1 receptors on cutaneous sensory nerves)
PART 4: CHOLESTATIC LIVER DISEASES
Cholestasis = impaired bile formation or flow. Divided into intrahepatic and extrahepatic.
4.1 Primary Biliary Cholangitis (PBC)
Previously called Primary Biliary Cirrhosis
Pathogenesis (Biochemical/Immunological):
- Autoimmune destruction of small intrahepatic bile ducts (interlobular bile ducts, <100 microns)
- Key autoantigen: E2 subunit of the pyruvate dehydrogenase complex (PDC-E2) in the inner mitochondrial membrane
- Anti-mitochondrial antibodies (AMA) directed against PDC-E2: present in 95% of PBC patients with sensitivity and specificity >95%
- Why do cholangiocytes become targets? PDC-E2 is aberrantly expressed on the apical surface of biliary epithelial cells in PBC patients → becomes accessible to immune attack
- T cell-mediated injury: PDC-E2-specific CD4+ and CD8+ T cells are present in periductal stroma
- Histologic hallmark: "Florid duct lesion" - lymphocytic/granulomatous destruction of bile ducts in portal tracts
- Additional antibodies: ANA, anti-sp100, anti-gp210 (nuclear pore proteins)
- Elevated serum IgM (characteristic but unexplained)
Metabolic/Biochemical Consequences of Progressive Cholestasis:
- Elevated ALP, GGT (markers of biliary epithelial injury)
- Elevated conjugated bilirubin (late disease)
- Hypercholesterolemia: Bile acid feedback on CYP7A1 is lost → cholesterol accumulates; also impaired biliary cholesterol excretion
- Xanthomas and xanthelasmas form (cholesterol deposits in skin/tendons)
- Paradoxically, cardiovascular risk is NOT increased because the excess cholesterol is in HDL
- Fat malabsorption: Reduced bile acid pool → impaired micelle formation → steatorrhea
- Fat-soluble vitamin deficiencies:
- Vitamin D deficiency → osteomalacia, osteoporosis
- Vitamin K deficiency → coagulopathy (elevated PT/INR)
- Vitamin A deficiency → night blindness
- Pruritus: Central mechanism involves excess bile acids and endogenous opioid accumulation; lysophosphatidic acid (LPA) generated by autotaxin (an enzyme upregulated in cholestasis) is the primary pruritogen
- Treatment: Ursodeoxycholic acid (UDCA) - hydrophilic bile acid that:
- Replaces toxic hydrophobic bile acids in the pool
- Has cytoprotective, anti-apoptotic, anti-inflammatory, choleretic effects
- Activates FXR and TGR5 signaling
- Obeticholic acid (OCA): Potent synthetic FXR agonist used in UDCA-refractory cases
4.2 Primary Sclerosing Cholangitis (PSC)
Characteristics:
- Inflammation and obliterative fibrosis of extrahepatic AND large intrahepatic bile ducts
- 70% male; median age 30; strongly associated with IBD (especially ulcerative colitis) in ~70% of cases
- Classic ERCP/MRCP: "string and bead pattern" - multifocal strictures with adjacent dilations ("beading"); "pruning" of smaller ducts
Pathogenesis:
- HLA associations: HLA-B8, DR3, DR4 → immune-mediated process
- Gut-liver axis hypothesis: In UC patients, T cells activated against gut antigens (possibly via molecular mimicry) migrate to liver and cross-react with bile duct antigens
- "Leaky gut" in IBD → bacterial products (LPS, secondary bile acids like DCA, LCA) reach liver via portal vein → trigger inflammatory injury to cholangiocytes
- Serological profile: pANCA positive (65%), AMA usually negative, IgG4 variable
- Biliary "bicarbonate umbrella" hypothesis: Normal cholangiocytes secrete HCO3- via AE2 anion exchanger, creating an alkaline layer that protects against toxic bile acids; in PSC, AE2 expression is reduced → cholangiocytes are exposed to toxic hydrophobic bile acids → apoptosis and injury
Biochemical Consequences:
- Elevated ALP, GGT disproportionately
- Progressive bile duct loss → impaired bile flow → secondary biliary cirrhosis
- Portal hypertension → varices, ascites, hepatic encephalopathy
- High risk of cholangiocarcinoma (~15% lifetime risk) - DNA damage from chronic bile acid exposure activates oncogenic pathways (KRAS, TP53, IDH1/2 mutations)
- No medical therapy proven to alter disease course (UDCA reduces ALP but no survival benefit demonstrated); liver transplantation is definitive
4.3 Secondary Sclerosing Cholangitis
Similar pattern to PSC but with a known cause:
- Post-surgical strictures
- Choledocholithiasis
- Ischemic cholangiopathy (ICU patients with prolonged hypotension)
- AIDS cholangiopathy (CMV, Cryptosporidium)
- IgG4-associated cholangitis (responds to steroids, unlike PSC)
PART 5: PROGRESSIVE FAMILIAL INTRAHEPATIC CHOLESTASIS (PFIC)
These are monogenic disorders of bile acid transport - among the most biochemically instructive bile disorders.
5.1 PFIC Type 1 - FIC1 Deficiency (Byler Disease)
- Gene: ATP8B1 (FIC1)
- Protein function: FIC1 is a P-type ATPase that acts as an aminophospholipid flippase, maintaining the asymmetric distribution of phospholipids in the canalicular membrane (phosphatidylserine on inner leaflet). This protects the membrane from the detergent action of bile salts.
- Without FIC1: Membrane integrity is lost → disrupted bile acid secretion → intrahepatic cholestasis
- Biochemistry: Low GGT (paradoxically - because phospholipid flipping defect impairs GGT activity), normal to low bile acids in bile, elevated bile acids in serum
- Presents in infancy/childhood with severe cholestasis, pruritus, growth failure
- Extra-hepatic manifestations: diarrhea (FIC1 is also expressed in intestine), hearing loss, pancreatitis
5.2 PFIC Type 2 - BSEP Deficiency
- Gene: ABCB11
- Protein function: Bile salt export pump (BSEP) - the primary ATP-dependent transporter secreting conjugated bile acids across the canalicular membrane into bile
- Without BSEP: Conjugated bile acids accumulate inside hepatocytes → direct hepatocellular toxicity → severe cholestasis → cirrhosis
- Biochemistry: Very low GGT, massively elevated serum bile acids (bile acid levels in serum can be 100x normal), elevated AST/ALT
- Most severe of the PFIC types; risk of hepatocellular carcinoma even in childhood
- BSEP is also inhibited by certain drugs (cyclosporine, rifampin, troglitazone) → drug-induced cholestasis
5.3 PFIC Type 3 - MDR3 Deficiency
- Gene: ABCB4
- Protein function: MDR3 is a phospholipid flippase (flips phosphatidylcholine/lecithin from inner to outer leaflet of canalicular membrane → lecithin is then secreted into bile)
- Lecithin in bile is critical: It forms mixed micelles with bile acids, preventing bile acids from acting as detergents on bile duct epithelium
- Without MDR3: Bile lacks lecithin → bile acids in unmicellized form directly damage cholangiocytes → biliary inflammation, sclerosing cholangitis-like picture
- Biochemistry: Elevated GGT (in contrast to PFIC 1 and 2!); bile is watery with very low phospholipid content; elevated serum ALP, GGT, conjugated bilirubin
- Adult presentation possible as "low-phospholipid-associated cholelithiasis (LPAC)" - recurrent cholesterol stones and biliary symptoms despite young age and absence of classical risk factors
- Treatment: UDCA (increases phospholipid secretion via alternative mechanisms)
PART 6: INHERITED DEFECTS IN BILE ACID SYNTHESIS
6.1 3beta-HSD Deficiency (HSD3B7)
- Most common inborn error of primary bile acid synthesis
- Cannot convert 3beta-hydroxy-Δ5 intermediates to the correct 3alpha-hydroxy configuration needed for CA and CDCA
- Atypical bile acids accumulate (3beta-hydroxy-Δ5 C27 bile acids) → toxic to hepatocytes → neonatal cholestasis
- Diagnosis: urine bile acid mass spectrometry shows characteristic atypical bile acids
- Treatment: oral CA/CDCA supplementation (replaces missing primary bile acids + suppresses CYP7A1 via FXR feedback)
6.2 Delta4-3-Oxosteroid 5beta-Reductase Deficiency (AKR1D1)
- Enzyme converts Δ4-3-oxo intermediates to 3-oxo-5beta configuration (required for correct sterol ring structure of bile acids)
- Accumulation of hepatotoxic Δ4-3-oxo bile acids
- Severe neonatal cholestasis and liver failure
- Treatment: primary bile acid replacement
6.3 CYP7A1 Deficiency
- Complete loss of the classical pathway
- Bile acid production reduced by ~90%
- Compensated by upregulation of the alternative pathway
- Adult presentation: hypercholesterolemia (cannot eliminate cholesterol as bile acids), gallstones
- Heterozygotes may have elevated LDL due to reduced CYP7A1 activity
6.4 Cerebrotendinous Xanthomatosis (CTX) - CYP27A1 Deficiency
- Gene: CYP27A1 (sterol 27-hydroxylase)
- Cannot hydroxylate the side chain of cholesterol → bile acid synthesis is defective (alternative pathway blocked)
- Accumulation of cholestanol (a reduced form of cholesterol) and bile alcohols in tissues
- Clinical features: Tendon xanthomas, cataracts, progressive neurological disease (cerebellar ataxia, dementia, spastic paraplegia), premature atherosclerosis
- Diagnosis: Elevated plasma cholestanol, elevated urinary bile alcohols
- Treatment: CDCA (suppresses CYP7A1 via FXR, reducing cholestanol synthesis) + statins
PART 7: DRUG-INDUCED CHOLESTASIS (BIOCHEMICAL MECHANISMS)
Many drugs disrupt bile formation through specific transporter inhibition:
| Drug | Mechanism | Effect |
|---|---|---|
| Cyclosporine | Inhibits BSEP (ABCB11) and MRP2 | Cholestasis |
| Rifampin | Inhibits NTCP, BSEP | Impaired bile acid uptake and secretion |
| Estrogens | Downregulate NTCP, BSEP expression; activate FXR → reduce CYP7A1 | Intrahepatic cholestasis of pregnancy |
| Troglitazone | Inhibits BSEP | Cholestasis (withdrawn from market) |
| Erythromycin | Inhibits MDR1/P-gp; may inhibit CYP3A4 → bile acid metabolite accumulation | Cholestatic hepatitis |
| Anabolic steroids (17alpha-alkylated) | Inhibit canalicular transporters (BSEP, MRP2) | "Pure" cholestasis |
Intrahepatic Cholestasis of Pregnancy (ICP):
- Late-pregnancy estrogen and progesterone surges inhibit BSEP and MRP2
- Bile acids accumulate in maternal plasma → cross placenta → fetal distress, stillbirth risk
- Genetic predisposition: many ICP patients carry heterozygous mutations in ABCB11 or ABCB4
- Treatment: UDCA (restores BSEP function, reduces fetal bile acid exposure)
PART 8: BILE ACID MALABSORPTION (BAM)
- Loss of terminal ileum (Crohn disease, resection) → bile acids not reabsorbed → spill into colon
- In colon: bile acids stimulate Cl-/water secretion (activate TGR5 on colonocytes → cAMP → CFTR activation) → secretory diarrhea
- Also: bile acids induce 7alpha-dehydroxylation by colonic bacteria → excess deoxycholic acid (hydrophobic) → mucosal damage, increased colorectal cancer risk
- Bile acid pool depletion → reduced micellar solubilization → fat malabsorption (steatorrhea) when >100 cm ileum lost
- SeHCAT test (75Se-homotaurocholic acid retention) - diagnostic test for BAM
- Treatment: Bile acid sequestrants (cholestyramine, colesevelam) to bind excess bile acids in colon; UDCA for mild cases
PART 9: BILE DUCT TUMORS
Cholangiocarcinoma
- Malignancy of bile duct epithelium (cholangiocytes)
- Risk factors all involve chronic bile acid exposure and inflammation:
- PSC, liver flukes (Clonorchis, Opisthorchis), hepatolithiasis, choledochal cysts
- Molecular biochemistry: Accumulation of lithocholic acid (secondary bile acid) promotes DNA damage (reactive oxygen species generation, NF-kB activation, AP-1 signaling); activates EGFR, VEGF pathways
- Key mutations: IDH1/2 (found in ~20% intrahepatic CC), FGFR2 fusions (~15% intrahepatic CC), KRAS, TP53, SMAD4
- Marker: CA 19-9 elevated (but not specific); CEA elevated
- IDH1/2 and FGFR2 are now therapeutic targets (ivosidenib for IDH1-mutant CC; pemigatinib, infigratinib for FGFR2-fusion CC)
Summary Reference Table: Key Biochemical Defects in Bile Disorders
| Disorder | Defective Molecule | Biochemical Consequence | Key Lab Finding |
|---|---|---|---|
| Gilbert syndrome | UGT1A1 (promoter) | Reduced bilirubin glucuronidation | Unconjugated bili↑, mildly |
| Crigler-Najjar I | UGT1A1 (absent) | No bilirubin conjugation | UCB >20 mg/dL, kernicterus |
| Dubin-Johnson | MRP2 (ABCC2) | Conjugated bili cannot exit hepatocyte | Conjugated bili↑, black liver |
| Rotor syndrome | OATP1B1/1B3 | Impaired hepatic conjugated bili reuptake | Conjugated bili↑, normal liver |
| Cholesterol gallstones | CSI >1, nucleation, stasis | Crystal/stone formation | Supersaturated bile |
| Pigment gallstones | Excess UCB in bile | Calcium bilirubinate precipitation | Hemolysis, infection |
| PBC | PDC-E2 autoantigen | Destruction of small ducts, cholestasis | AMA+, ALP↑, IgM↑ |
| PSC | Cholangiocyte injury | Fibro-obliterative large duct disease | pANCA+, ALP↑, "beading" on MRCP |
| PFIC 1 (FIC1) | ATP8B1 | Membrane integrity loss | Low GGT, cholestasis |
| PFIC 2 (BSEP) | ABCB11 | No canalicular bile acid export | Very low GGT, extreme BA↑ |
| PFIC 3 (MDR3) | ABCB4 | No lecithin in bile → detergent injury | High GGT, low biliary phospholipid |
| CTX | CYP27A1 | Cholestanol accumulation | Elevated plasma cholestanol |
| Bile acid malabsorption | ASBT or ileal loss | Bile acids spill into colon | SeHCAT <10% retention |
| ICP | BSEP/ABCB11 (estrogen effect) | Bile acid retention in pregnancy | Bile acids↑, pruritus |
Sources:
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease - Chapter 64 (Bile Acid Synthesis), Chapter 77 (Inborn Errors), Chapter 21 (Differential Diagnosis of Jaundice)
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Chapter 18 (PBC, PSC)
- Robbins & Kumar Basic Pathology - Chapter 14 (Cholelithiasis)
- Harrison's Principles of Internal Medicine 22E - Gallstones chapter
Explain biochemistry of bile secretions
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Loading Image
Reading File
Finding Sources
Loading Image
Reading File
I now have comprehensive, highly detailed material from the primary gastroenterology textbooks. Let me compose the full response.
Biochemistry of Bile Secretion
Overview
Bile secretion is not a simple filtration process - it is an active, energy-driven, multi-step process of organic and inorganic solute secretion that creates an osmotic gradient drawing water along behind it. The final bile is nearly isotonic with plasma (~300 mOsmol/kg), yet highly concentrated in organic molecules. Two cell types contribute: hepatocytes (produce primary canalicular bile) and cholangiocytes (modify and augment it in the bile ducts).
Total daily bile secretion: 500-1500 mL/day (hepatocytes contribute ~80%, cholangiocytes ~20%)
PART 1: Composition of Bile
1.1 Hepatic Bile (freshly secreted)
| Component | % by moles | Function |
|---|---|---|
| Bile acids | 80% | Micelle formation, fat digestion, driving force for bile flow |
| Phospholipids (lecithin/phosphatidylcholine, cephalins, sphingomyelin) | 16% | Co-micellar solubilization of cholesterol; protect bile ducts from bile acid detergency |
| Unesterified cholesterol | 4% (up to 8-10% in lithogenic states) | Excreted; if excess, causes gallstones |
| Conjugated bilirubin | Minor | Excretory waste product of heme catabolism |
| Proteins | Minor | Secretory IgA, albumin, hormone metabolites, enzymes |
| Electrolytes | Isotonic with plasma | Na+, K+, Cl-, HCO3- |
| Mucus glycoproteins | Trace | Lubrication; can be nucleating factor for gallstones |
| Glutathione (GSH) | 1-4 mM | Drives bile acid-independent bile flow |
| Heavy metals, drugs, drug metabolites | Variable | Biliary excretion route |
1.2 Gallbladder Bile (after concentration)
The gallbladder actively reabsorbs water and inorganic electrolytes (NaCl, NaHCO3), concentrating the organic solutes 5-10x:
- Total solute concentration rises from 3-4 g/dL (hepatic bile) to 10-15 g/dL (gallbladder bile)
- Na+, Cl-, and HCO3- are greatly reduced (absorbed)
- Organic solutes (bile acids, phospholipids, bilirubin, cholesterol) are concentrated proportionally
- pH falls (from ~7.4 to ~6-7) due to bicarbonate reabsorption and proton secretion
PART 2: The Two Fundamental Mechanisms of Bile Formation
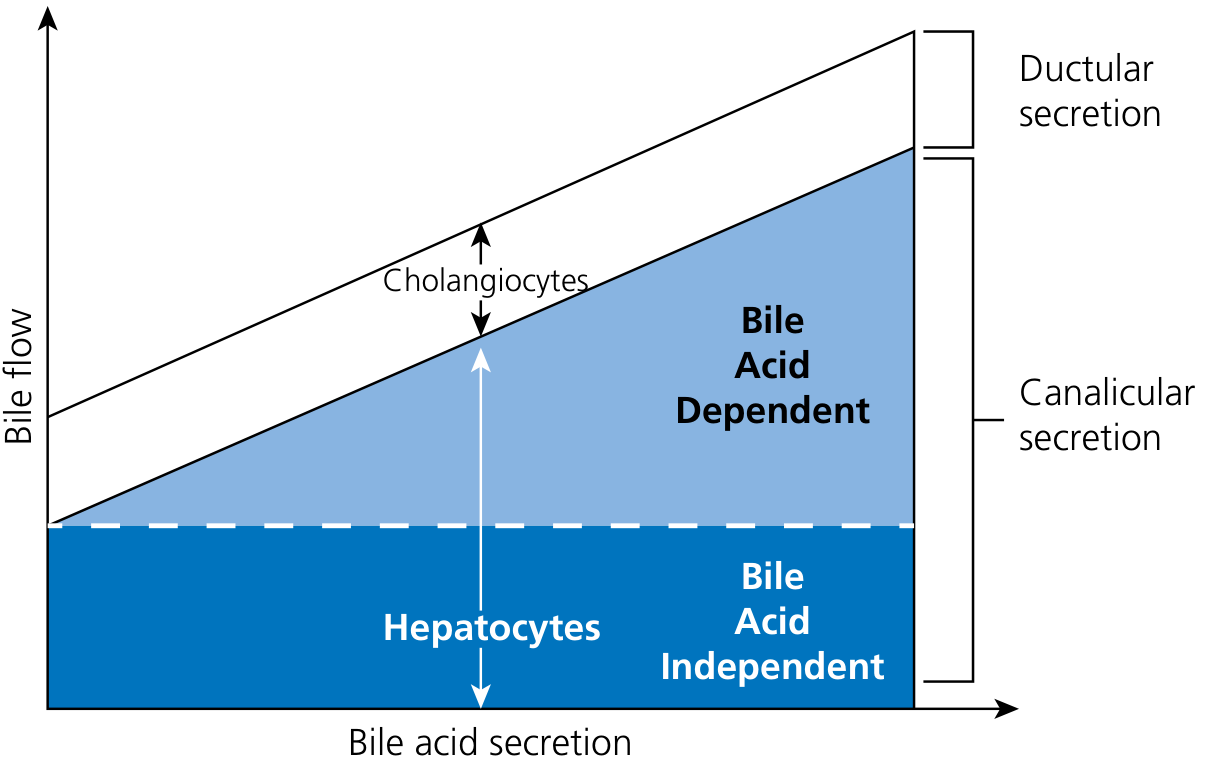
Canalicular bile flow is classified into two components:
2.1 Bile Acid-Dependent Bile Flow (BADBF)
- Driven by the osmotic effects of bile acids concentrated in the canalicular lumen
- As bile acids are actively pumped into the bile canaliculus by BSEP (see Part 3), they create a high-osmolality zone
- Water follows osmotically through aquaporin channels (AQP8) on the canalicular membrane and via paracellular junctions (tight junctions between adjacent hepatocytes)
- Electrolytes (Na+, K+, Cl-) follow passively
- Each 1 µmol of taurocholate secretion produces ~8 µL of additional bile flow (from both BADBF and stimulation of ductular HCO3-/Cl- secretion)
- Bile flow correlates linearly with bile acid secretion rate
2.2 Bile Acid-Independent Bile Flow (BAIBF)
- Even with complete absence of bile acids, ~0.8-1.25 µL/min/g liver of bile flow persists
- Driven by secretion of two principal anions into the canaliculus:
a) Glutathione (GSH):
- Secreted at 4-9 mM/min/g liver via MRP2/ABCC2 at the canalicular membrane
- GSH concentration in bile: 1-4 mM
- Once in bile, each GSH molecule is cleaved by gamma-glutamyl transpeptidase (GGT) on the canalicular surface: GSH → glutamate + cysteinyl-glycine
- The amino acid fragments act as osmotic drivers
- Direct correlation between GSH secretion and BAIBF
b) Bicarbonate (HCO3-):
- Secreted into the canalicular lumen via the AE2 (anion exchanger 2 / SLC4A2) Cl-/HCO3- exchanger on the canalicular membrane
- HCO3- secretion creates an osmotic gradient for water entry
- Creates the "biliary bicarbonate umbrella" - an alkaline layer protecting the canalicular/ductal epithelium from the detergent/acid action of bile acids
PART 3: Hepatocyte Transport Biochemistry - Step by Step
Bile secretion by hepatocytes is a vectorial (directional) transport process: solutes move from sinusoidal blood → hepatocyte cytoplasm → bile canaliculus. The polarized hepatocyte has distinct membrane domains for each step.
Step 1 - Sinusoidal (Basolateral) Uptake
The energy master key: Na+/K+-ATPase (on basolateral membrane)
- Pumps 3 Na+ OUT and 2 K+ IN per cycle (using ATP)
- Creates: (1) inwardly directed Na+ chemical gradient; (2) inside-negative electrical potential of -35 to -40 mV
- These two driving forces are harnessed by all Na+-coupled cotransporters
Bile acid uptake:
- NTCP (Na+-taurocholate cotransporting polypeptide, SLC10A1): Primary transporter for conjugated bile acids. Cotransports 2 Na+ with 1 bile acid anion inward - uses both Na+ chemical gradient AND membrane potential. Achieves 40-fold concentration of bile acids inside hepatocyte vs. portal blood.
- OATP1B1 (SLCO1B1) and OATP1B3 (SLCO1B3): Na+-independent uptake - use the inside-negative membrane potential. Transport unconjugated bile acids, bilirubin-albumin complexes, drugs (statins, rifampin), and other organic anions. Critical pharmacological drug uptake transporters.
Bilirubin uptake:
- Unconjugated bilirubin is bound to albumin in plasma
- Dissociates at the sinusoidal membrane; bilirubin enters hepatocyte either by facilitated diffusion or via OATPs
- Intracellularly, bound to ligandin (Y-protein / glutathione-S-transferase) - prevents it from diffusing back out
Other substrates:
- Free fatty acids → bound to liver fatty acid-binding protein (L-FABP)
- Organic cations → via OCT (organic cation transporters)
- Drugs, hormones → via OATPs
Step 2 - Intracellular Transport
Two mechanisms carry solutes from sinusoidal membrane to canalicular membrane:
a) Protein/cytosol-mediated transport:
- Small lipophilic molecules (bile acids, fatty acids, bilirubin) are bound to intracellular binding proteins and carried across
- Bile acids are delivered to the Golgi apparatus, loaded into vesicles, and transported to the canalicular membrane
b) Vesicular transcytotic transport:
- Large proteins (secretory IgA, transferrin) bind specific sinusoidal receptors → undergo receptor-mediated endocytosis → vesicles traffic through the cytoplasm via microtubules → exocytosis at the canalicular membrane
- Hepatocytes maintain an extraordinarily high rate of membrane turnover (~5× entire plasma membrane area per hour), reflecting the intensity of vesicular trafficking
Bilirubin conjugation (occurs in the endoplasmic reticulum):
- Free bilirubin in hepatocyte cytoplasm → ER
- UGT1A1 (UDP-glucuronosyltransferase 1A1): transfers glucuronate from UDP-glucuronate to bilirubin
- Products: bilirubin monoglucuronide and bilirubin diglucuronide (the major form excreted)
- Conjugation renders bilirubin water-soluble for canalicular excretion
Bile acid conjugation (amino acid amidation):
- Primary bile acids (CA, CDCA) are activated: bile acid-CoA ligase (BACL1/SLC27A5) forms bile acid-CoA thioester
- BAAT (bile acid-CoA:amino acid N-acyltransferase) conjugates the CoA thioester with glycine or taurine
- Glycine conjugates (glycocholate, glycochenodeoxycholate) predominate (ratio glycine:taurine = ~3:1 in humans)
- Purpose: Lowers pKa (from ~6 to ~2 for taurine; ~4 for glycine) → fully ionized at intestinal pH → stays in lumen, not passively absorbed → maintained in enterohepatic pool
Step 3 - Canalicular (Apical) Secretion
This is the rate-limiting step for bile formation. The canalicular membrane is packed with ATP-binding cassette (ABC) transporters that pump solutes against enormous concentration gradients using ATP hydrolysis.
| Transporter | Gene | Substrate | Disease if absent |
|---|---|---|---|
| BSEP (Bile Salt Export Pump) | ABCB11 | Conjugated bile acids | PFIC-2, BRIC-2, ICP |
| MDR3 (Multidrug Resistance 3) | ABCB4 | Phosphatidylcholine (lecithin) | PFIC-3, low-phospholipid cholelithiasis |
| ABCG5/G8 (heterodimer) | ABCG5, ABCG8 | Cholesterol, plant sterols | Beta-sitosterolemia (absent); excess cholesterol in bile (overactive) |
| MRP2 (Multidrug Resistance-associated Protein 2) | ABCC2 | Conjugated bilirubin, glutathione, drug glucuronides/sulfates, sulfated bile acids | Dubin-Johnson syndrome |
| MDR1 (P-glycoprotein) | ABCB1 | Hydrophobic organic cations, xenobiotics, some drugs | Drug resistance (no human cholestasis described) |
| BCRP (Breast Cancer Resistance Protein) | ABCG2 | Sulfated conjugates, hormones, drugs | Drug resistance |
| FIC1 (Familial Intrahepatic Cholestasis 1) | ATP8B1 | Aminophospholipids (flippase) | PFIC-1, BRIC-1 |
ATP-independent canalicular transporters:
- AE2 (SLC4A2): Cl-/HCO3- anion exchanger → secretes HCO3- into bile (biliary bicarbonate umbrella)
- AQP8 (Aquaporin 8): Water channel on canalicular membrane; allows osmotically-driven water entry
Canalicular membrane integrity (FIC1/ATP8B1):
- The canalicular membrane is exposed to highly concentrated bile acids (potential detergent injury)
- FIC1 is a flippase that maintains the asymmetric distribution of aminophospholipids (phosphatidylserine and phosphatidylethanolamine confined to the inner leaflet)
- This lipid asymmetry is essential for membrane stability and the function of all other ABC transporters
- Loss of FIC1 → ablation of ATP-dependent transporter function → cholestasis
PART 4: Ductular Bile Secretion - Cholangiocyte Biochemistry
After leaving hepatocyte canaliculi, bile enters bile ductules and then large bile ducts lined by cholangiocytes. Cholangiocytes add ~20% of total bile volume and critically modify bile composition.
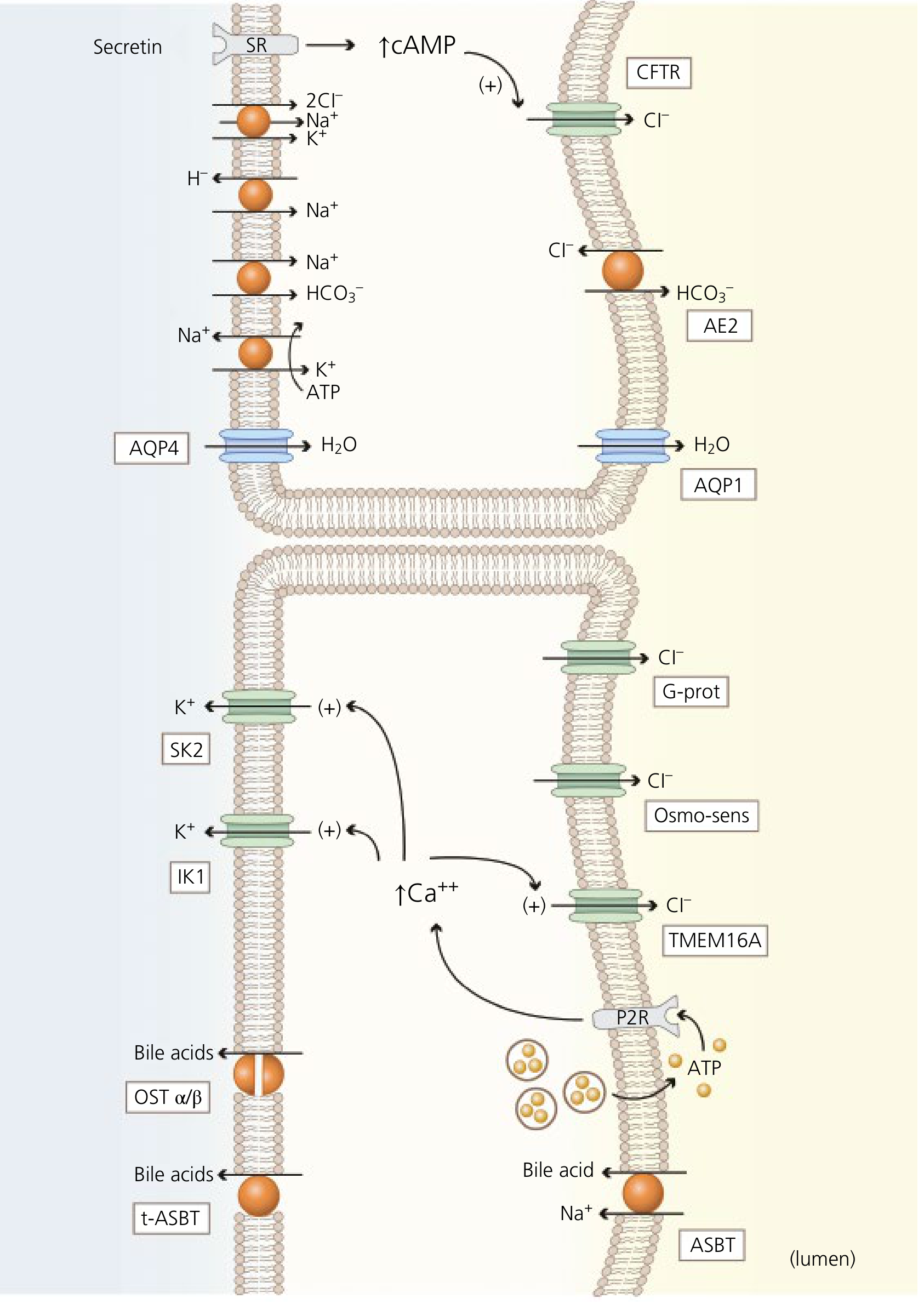
The Secretin-cAMP-CFTR-AE2 Cascade (Choleresis)
This is the dominant mechanism of ductular bile secretion:
Step 1 - Secretin stimulation:
- Duodenal S-cells release secretin in response to luminal acid (H+) from gastric emptying
- Secretin binds secretin receptor (SR) on the basolateral membrane of cholangiocytes (medium and large cholangiocytes only - small cholangiocytes lack SR)
Step 2 - cAMP generation:
- SR is coupled to Gs protein → activates adenylyl cyclase → increases intracellular cAMP
- cAMP activates Protein Kinase A (PKA)
Step 3 - CFTR activation:
- PKA phosphorylates CFTR (cystic fibrosis transmembrane regulator) on the apical membrane
- CFTR opens → Cl- efflux into the bile duct lumen
- A lumen-negative electrical potential is created
Step 4 - AE2 activation:
- Luminal Cl- accumulation + lumen-negative potential drives AE2 (Cl-/HCO3- exchanger): exchanges lumen Cl- back for intracellular HCO3- → net HCO3- secretion into bile
- This alkalinizes and dilutes bile
Step 5 - Water secretion:
- The osmotic gradient created by Cl- and HCO3- drives water through:
- AQP1 (aquaporin 1) on the apical (luminal) membrane
- AQP4 (aquaporin 4) on the basolateral membrane
Supporting ion transport at basolateral membrane (maintains HCO3- supply and intracellular pH):
- Na+/K+/2Cl- cotransporter (NKCC1): Loads Cl- into cell for apical secretion
- Na+/H+ exchanger (NHE): Expels H+ to maintain intracellular pH
- Na+/HCO3- cotransporter (NBC): Imports HCO3- from blood
- K+ channels (SK2, IK1): K+ efflux hyperpolarizes the membrane → maintains driving force for continued Cl- secretion
Secondary Cl- channels in cholangiocytes:
- TMEM16A: Ca2+-activated Cl- channel; activated by intracellular Ca2+ increases (triggered by nucleotides, bile acids acting on apical P2Y receptors)
- LRRC8A: Volume-sensitive (osmo-sensitive) Cl- channel; activated during cell swelling
- G-protein regulated Cl- channel: Basolateral; role uncertain
Cystic Fibrosis and bile:
- CFTR mutations → absent/dysfunctional CFTR in cholangiocytes → no Cl- efflux → no Cl-/HCO3- exchange → inspissated, dehydrated bile → bile duct obstruction → biliary cirrhosis in CF patients
Cholangiocyte Bile Acid Absorption (Cholehepatic Shunting)
Cholangiocytes of the large bile ducts also express ASBT (apical Na+-dependent bile acid transporter, SLC10A2) on their luminal membrane:
- Reabsorbs some bile acids from bile duct lumen back into cholangiocytes
- Bile acids exit basolaterally via OSTalpha-OSTbeta heterodimer and t-ASBT (truncated ASBT)
- Re-enter hepatic arterial blood → return to hepatocytes → re-secreted into bile
- This "cholehepatic shunting" allows a portion of the bile acid pool to recirculate directly within the liver, bypassing the full enterohepatic circulation
PART 5: Regulation of Bile Secretion
5.1 Hormonal Regulation
| Hormone | Source | Receptor | Mechanism | Effect on Bile |
|---|---|---|---|---|
| Secretin | Duodenal S-cells (response to H+) | SR on cholangiocytes (basolateral) | cAMP → PKA → CFTR → AE2 | Increases HCO3--rich ductular secretion; choleresis |
| Cholecystokinin (CCK) | Duodenal I-cells (response to fats, proteins) | CCK-A receptor on gallbladder smooth muscle | IP3/Ca2+ → smooth muscle contraction | Gallbladder contraction + sphincter of Oddi relaxation → bile delivery to duodenum |
| Glucagon | Pancreatic alpha-cells | Gs-coupled receptor | cAMP increase | Mild choleresis |
| Vasoactive Intestinal Peptide (VIP) | ENS neurons | VPAC receptor | cAMP increase | Choleresis; relaxes sphincter of Oddi |
| Motilin | Duodenal M-cells | Motilin receptor | Smooth muscle contraction | Gallbladder contraction during fasting (migrating motor complex) |
| Somatostatin | D-cells | Gi-coupled receptor | cAMP decrease | Inhibits bile secretion and gallbladder contraction |
| FGF19 | Ileal enterocytes (FXR-induced) | FGFR4/beta-klotho on hepatocytes | Represses CYP7A1 | Reduces bile acid synthesis; promotes gallbladder relaxation (filling) during fasting |
5.2 Neural Regulation
- Parasympathetic (vagus nerve): ACh → M3 muscarinic receptors on hepatocytes and gallbladder → increases bile secretion and gallbladder contraction
- Sympathetic (adrenergic): Inhibits bile secretion; causes sphincter of Oddi contraction
- Vagal activity during the cephalic phase (sight/smell of food) pre-emptively increases bile flow before food reaches the duodenum
5.3 Nuclear Receptor Regulation (Transcriptional)
Nuclear receptors are ligand-activated transcription factors that coordinate the expression of all bile secretion genes. The most important:
| Receptor | Key Ligands | Genes Activated | Genes Repressed | Net Effect |
|---|---|---|---|---|
| FXR (NR1H4) | Bile acids (CDCA > DCA > CA > LCA) | BSEP, MRP2, MDR3, OSTalpha/beta, SHP, PXR, CYP3A4 | NTCP, CYP7A1 | Master bile acid sensor: increases secretion, decreases synthesis and uptake |
| SHP (NR0B2) | (Induced by FXR) | - | CYP7A1, CYP8B1, NTCP, ASBT | Secondary negative regulator; prevents bile acid overload |
| PXR (NR1I2) | Xenobiotics, rifampin, LCA, statins | CYP3A4, MDR1, MRP2, UGT1A1 | CYP7A1 | Detoxification; especially important for handling toxic secondary bile acids (LCA) |
| CAR (NR1I3) | Phenobarbital, bilirubin, xenobiotics | CYP3A, CYP2B6, MRP2, UGT1A1 | - | Drug and bilirubin detoxification; activated by bilirubin accumulation |
| LXRalpha (NR1H3) | Oxysterols, 6alpha-hydroxy bile acids | ABCG5/G8, ABCA1, CYP7A1 | - | Cholesterol export into bile; upregulates bile acid synthesis when cholesterol is high |
| VDR (NR1I1) | Vitamin D, LCA | CYP3A4, SULT2A1 | - | Detoxification of toxic LCA via hydroxylation and sulfation |
| HNF4alpha (NR2A1) | - | CYP7A1, CYP8B1, NTCP | - | Positive regulator of bile acid synthesis and uptake |
| RXR | 9-cis-retinoic acid | (Heterodimerization partner) | - | Required co-receptor for FXR, PXR, LXR, CAR; cytokines inhibit it |
Clinical example - Rifampin paradox:
- Short-term rifampin activates PXR → induces CYP3A4 and UGT1A1 → bile acid hydroxylation and detoxification → anti-cholestatic effect (relieves pruritus)
- Long-term or high-dose rifampin inhibits NTCP and BSEP directly → reduces bile acid transport → worsens cholestasis
PART 6: Gallbladder Function in Bile Secretion
The gallbladder acts as a reservoir and concentrating organ:
6.1 Storage and Concentration
During fasting (interdigestive period):
- Sphincter of Oddi contracts
- Gallbladder relaxes (FGF19 promotes relaxation via TGR5)
- Bile secreted by liver is diverted into the gallbladder
- Gallbladder epithelium actively absorbs: Na+, Cl-, HCO3-, and water (following osmotic gradient)
- Na+ absorbed via apical Na+/H+ exchanger and Na+/Cl- cotransporter
- Cl- absorbed via Cl-/HCO3- exchanger and Cl- channels
- Water follows via AQP1 and paracellular routes
- Result: 5-10x concentration of organic solutes (bile acids, bilirubin, cholesterol, phospholipids)
6.2 Gallbladder Mucin Secretion
- Gallbladder epithelium secretes mucin glycoproteins into bile
- Mucin lubricates the biliary tree
- Pathologically, mucin hypersecretion (stimulated by lithogenic bile) forms a gel that traps cholesterol crystals → promotes gallstone nucleation and growth
6.3 Gallbladder Emptying
During a meal (postprandial phase):
- Dietary fats and proteins in the duodenum → stimulate I-cells → release CCK
- CCK binds CCK-A receptors on gallbladder smooth muscle → IP3/DAG pathway → intracellular Ca2+ rise → smooth muscle contraction
- Simultaneously: CCK relaxes sphincter of Oddi (via VIP release from enteric neurons)
- Vagal stimulation (parasympathetic) augments gallbladder contraction
- Concentrated bile flows from gallbladder → cystic duct → common bile duct → sphincter of Oddi → duodenum
- Emptying fraction: ~75% per meal in normal subjects
- When gallbladder is absent (post-cholecystectomy): bile is stored in the proximal small intestine during fasting; pulsatile flow is maintained by the migrating motor complex; after a meal, small intestinal contractions deliver bile acids to the terminal ileum for reabsorption
PART 7: Bile Acid Function in the Intestinal Lumen
Once secreted into the duodenum, bile acids perform critical digestive biochemistry:
7.1 Micellar Solubilization
- Above their critical micellar concentration (CMC ~1.5-2 mM), bile acid molecules spontaneously aggregate into micelles
- Bile acid molecules are amphipathic: hydrophilic (hydroxyl groups) face outward; hydrophobic (sterol ring) faces inward
- Mixed micelles also incorporate lecithin (phosphatidylcholine): form larger, more effective solubilization units
- Mixed micelles solubilize: monoglycerides, fatty acids, fat-soluble vitamins (A, D, E, K), cholesterol, and other lipids
- Without micelles, lipid digestion products cannot cross the unstirred water layer to reach enterocyte brush border
7.2 Activation of Pancreatic Lipase
- Bile salts displace colipase from the oil-water interface of triglyceride droplets
- Colipase-pancreatic lipase complex then anchors to the surface → efficient triglyceride hydrolysis → monoglycerides + fatty acids
7.3 Electrolyte and Water Absorption
- Physiologic bile acid concentrations stimulate Na+ and water absorption in the small bowel
- In the colon at pathological concentrations (bile acid malabsorption): activate TGR5 → cAMP → Cl- secretion → secretory diarrhea (bile acid-induced diarrhea)
7.4 Bile Acid Signaling (Hormone-like Actions)
Bile acids are now recognized as systemic hormones acting via:
- FXR (nuclear receptor): In ileal enterocytes, liver, and other tissues; regulates bile acid metabolism, glucose homeostasis, lipid metabolism
- TGR5 (G-protein-coupled receptor): On cholangiocytes, gallbladder epithelium, enteroendocrine cells, brown adipose tissue, muscle; regulates gallbladder filling, gut motility, GLP-1 secretion (incretin effect), energy expenditure
- S1PR2 (sphingosine-1-phosphate receptor 2): On hepatocytes; activates NF-kB, regulates hepatic lipid metabolism
PART 8: Enterohepatic Circulation - The Complete Cycle
Liver (hepatocytes)
↓ BSEP secretes conjugated bile acids into canaliculi
↓ bile flows through canaliculi → ductules → bile ducts
↓ (cholangiocyte modification - HCO3-, water added)
↓ sphincter of Oddi relaxes (CCK)
Duodenum
↓ mixed micelle formation, fat digestion
↓ passive absorption along entire small intestine (unconjugated, some conjugated)
Terminal ileum
↓ ACTIVE uptake via ASBT (SLC10A2) - conjugated bile acids
↓ EXIT via OSTalpha/OSTbeta at basolateral membrane
Portal blood
↓ bile acids bound to albumin travel to liver
Hepatocyte (sinusoidal membrane)
↓ NTCP re-uptakes conjugated bile acids (Na+-coupled)
↓ OATPs uptake unconjugated bile acids
↓ Reconjugation with glycine/taurine
↓ Re-secretion via BSEP → cycle repeats
Efficiency metrics:
- Pool recycles 5-10 times/day
- 10-30 g bile acids reabsorbed/day from 2-4 g pool
- Fecal loss: 0.2-0.4 g/day (replaced by de novo synthesis from cholesterol)
- The maximum synthetic capacity is ~5 g/day; if intestinal losses exceed this (e.g., massive ileal resection), the pool cannot be replenished
Summary: Key Biochemical Cascade
CHOLESTEROL (hepatocyte)
↓ CYP7A1 (rate-limiting; inhibited by FXR-FGF19)
PRIMARY BILE ACIDS (Cholic acid, Chenodeoxycholic acid)
↓ BAAT (conjugation with glycine/taurine)
CONJUGATED PRIMARY BILE SALTS
↓ BSEP (ATP-dependent, canalicular)
BILE CANALICULUS (+ phospholipids via MDR3, cholesterol via ABCG5/G8,
bilirubin-glucuronide via MRP2, HCO3- via AE2,
GSH via MRP2)
↓ Cholangiocyte modification (Secretin→cAMP→CFTR→AE2: +HCO3-, +H2O)
BILE DUCT BILE
↓ Gallbladder concentration (NaCl/H2O absorbed, mucin added)
GALLBLADDER BILE
↓ CCK-stimulated contraction (meal)
DUODENUM: micelle formation → fat digestion → fat-soluble vitamin absorption
↓ Ileal ASBT active reabsorption (terminal ileum)
PORTAL BLOOD → HEPATOCYTE NTCP → Reconjugation → Re-secretion via BSEP
(Enterohepatic cycle: 5-10×/day)
Sources:
- Yamada's Textbook of Gastroenterology, 7th Ed. - Chapter 19 (Mechanisms of Bile Formation), pp. 435-445
- Harrison's Principles of Internal Medicine 22E - Bile Secretion and Composition section
- Sabiston Textbook of Surgery - Bile Formation chapter, pp. 1574-1582
- Sleisenger and Fordtran's GI and Liver Disease - Chapter 64 (Transport proteins, enterohepatic circulation)
This is a shared conversation. Sign in to Orris to start your own chat.