Pathogenesis of h pylori infecton
Reading File
Finding Sources
Searching PubMed
"Helicobacter pylori" AND pathogenesis
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive content from multiple authoritative textbooks with excellent diagrams. Let me compile the full detailed answer.
Pathogenesis of Helicobacter pylori Infection
False-colored scanning electron micrograph showing H. pylori (red) on gastric epithelial cells. - Yamada's Textbook of Gastroenterology, 7th ed.
1. The Organism
H. pylori is a gram-negative, spiral-shaped bacillus (~3.5 µm long, 0.5 µm wide). Its helical shape enables it to corkscrew through the thick gastric mucus layer. It is microaerophilic, urease-positive, and lives exclusively on gastric-type mucosa, deep in the mucus layer overlying the epithelium. - Yamada's Textbook of Gastroenterology, 7th ed.
2. Colonization and Survival in the Gastric Niche
Before causing disease, the organism must overcome the hostile environment of the stomach:
| Mechanism | How it works |
|---|---|
| Urease | Hydrolyzes urea → NH₃ + CO₂; ammonia buffers local acidity, maintaining periplasmic pH near neutral even in acid |
| Flagella | Corkscrew motility allows movement through viscous mucus; bacteria also sense pH and actively migrate away from acidic microenvironments toward the epithelial surface |
| Adhesins (outer membrane proteins) | BabA binds fucosylated Lewis-b (Le^b) antigen on epithelial cells; SabA binds sialyl-Lewis^x; HopQ binds CEACAM family molecules - anchoring the organism to the gastric epithelium and preventing clearance by peristalsis |
| Mucin degradation | H. pylori decreases mucin synthesis, helping it penetrate the mucous layer |
- Robbins, Cotran & Kumar Pathologic Basis of Disease; Yamada's Textbook of Gastroenterology, 7th ed.
3. Key Virulence Factors
A. CagA (Cytotoxin-Associated Gene A) - the "oncoprotein"
- Encoded within the cag pathogenicity island (cag PAI), a ~40-kb genomic segment
- CagA is injected directly into gastric epithelial cells via a type IV secretion system (T4SS) - essentially a molecular syringe
- Once inside host cells, CagA is tyrosine-phosphorylated by Src and Abl kinases, and activates multiple growth factor signaling pathways (Ras-ERK, PI3K, SHP-2)
- This stimulates abnormal cell proliferation, disrupts tight junctions, and contributes to the carcinogenic cascade
- CagA-positive strains cause more severe inflammation and are strongly associated with gastric adenocarcinoma and duodenal ulcer
- The cag T4SS also translocates heptose bisphosphate (HBP), a metabolite that activates NF-κB via TIFA, amplifying inflammatory cytokine production (IL-8, TNF-α, IL-1β)
- H. pylori DNA itself is translocated into host cells via T4SS, activating intracellular TLR9
B. VacA (Vacuolating Cytotoxin A)
- A secreted pore-forming toxin present in virtually all H. pylori strains, but with major functional variation based on allele type
- The vacA gene has polymorphic regions: the signal (s) region (s1 or s2) and the middle (m) region (m1 or m2)
- vacA s1/m1 strains induce strongest vacuolation and are most strongly linked to ulcer disease and gastric cancer
- s2/m2 strains show no vacuolating activity
- VacA effects on host cells:
- Induces large cytoplasmic vacuoles by disrupting endosomal/lysosomal trafficking
- Triggers apoptosis of epithelial cells
- Suppresses T-cell responses - contributing to the longevity of infection by impairing immune clearance
C. Other Virulence Factors
-
OipA (outer inflammatory protein A): induces IL-8 production
-
DupA: associated with duodenal ulcer risk
-
Lipopolysaccharide (LPS): weakly immunogenic compared to other gram-negatives; contains Lewis antigens (Le^x, Le^y) that mimic host antigens - a mechanism of molecular mimicry allowing immune evasion
-
Mulholland and Greenfield's Surgery, 7th ed.; Yamada's Textbook of Gastroenterology, 7th ed.; Sherris & Ryan's Medical Microbiology, 8th ed.
4. Host Inflammatory Response
All H. pylori infections are associated with histological gastritis - infiltration of lymphocytes and neutrophils into the gastric mucosa. The inflammatory cascade proceeds as follows:
- Bacterial adhesion and virulence factor injection trigger epithelial cells to secrete IL-8 and other chemokines
- Neutrophil and lymphocyte recruitment → mucosal infiltration
- Activated macrophages and T cells release TNF-α, IL-1β, IL-6, IL-12
- Reactive oxygen species (ROS) from neutrophils cause direct epithelial DNA damage
- Chronic inflammation drives polyclonal B-cell proliferation → can give rise to gastric MALToma if driver mutations are acquired
The pattern of gastritis (antral-predominant vs. pangastritis) is the key determinant of which disease the patient develops.
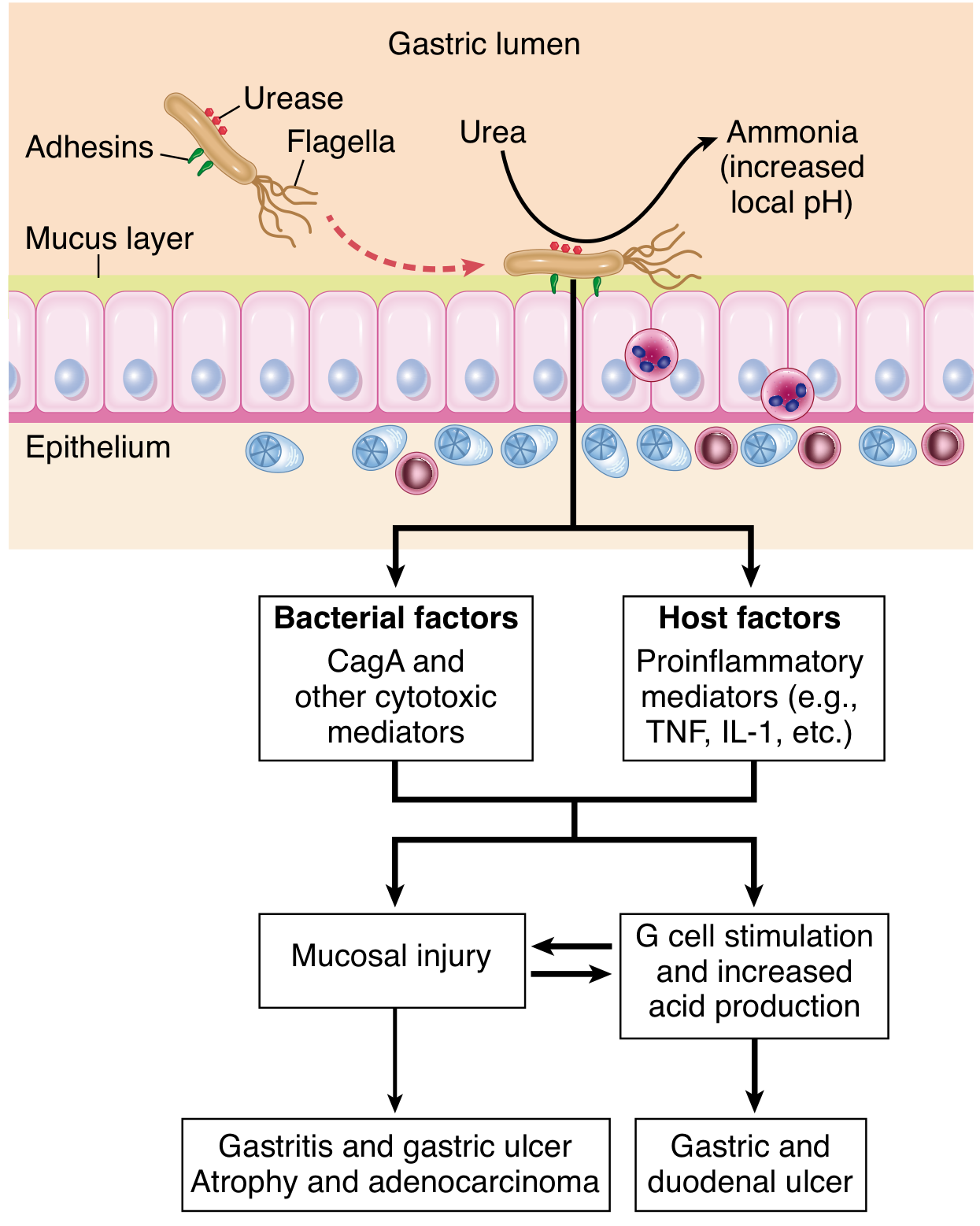
Pathogenesis of H. pylori gastritis. - Robbins, Cotran & Kumar Pathologic Basis of Disease
5. Disease Outcomes - Determined by Gastritis Pattern
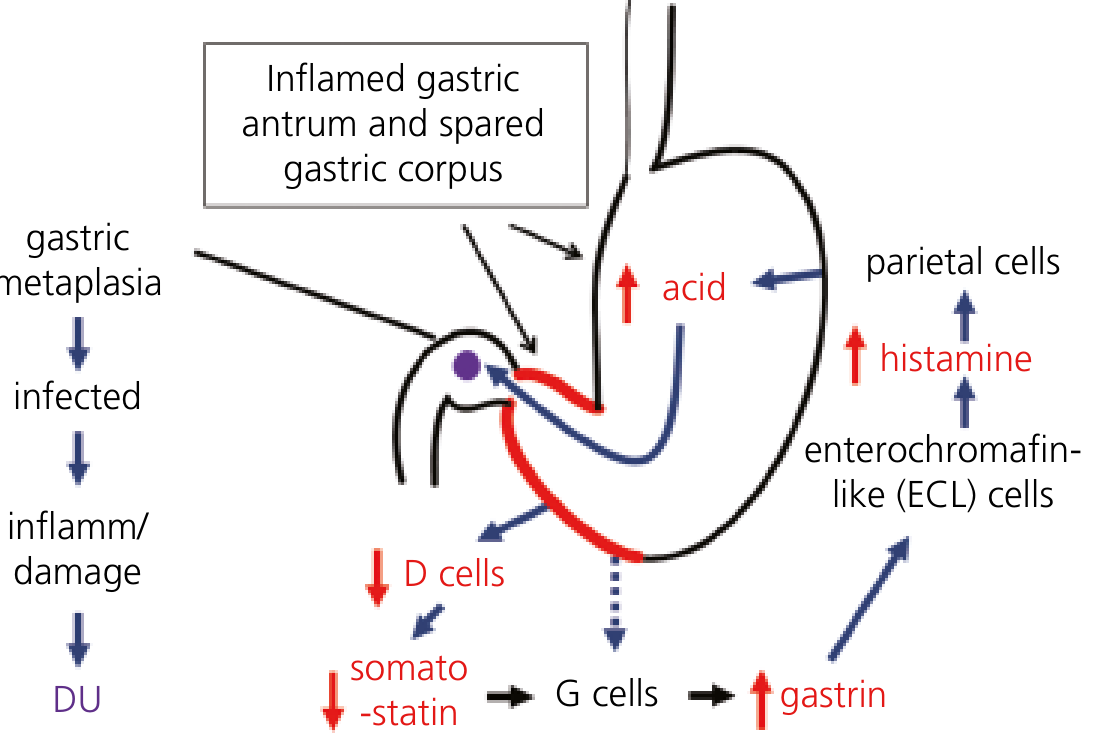
H. pylori-induced duodenal ulceration pathway: antral inflammation → ↓ somatostatin → ↑ gastrin → ↑ acid → duodenal metaplasia → DU. - Yamada's Textbook of Gastroenterology, 7th ed.
A. Antral-Predominant Gastritis → Duodenal Ulcer (DU)
- H. pylori colonizes predominantly the antrum, leaving the acid-producing corpus relatively spared
- Antral inflammation suppresses somatostatin production by D cells
- Loss of somatostatin removes the normal negative feedback on G cells → hypergastrinemia
- Excess gastrin increases parietal cell mass and stimulates ECL cells → ↑ histamine → ↑ acid output from the healthy corpus
- The increased acid load in the duodenum promotes gastric metaplasia of the duodenal bulb (protective response to excess acid)
- H. pylori can only colonize gastric-type mucosa - gastric metaplasia now allows duodenal colonization → local inflammation and ulceration
- Bicarbonate secretion by the duodenum is also reduced, removing a key mucosal defense
B. Pangastritis → Gastric Ulcer (GU) and Gastric Adenocarcinoma
- Inflammation involving both antrum and corpus (pangastritis)
- Corpus inflammation damages parietal cells → normal or reduced acid (so DU does not develop)
- Progressive atrophy of parietal glands → hypochlorhydria → continued hypergastrinemia
- Reduced acid creates a permissive environment for further bacterial colonization and mucosal damage
- If inflammation is severe, particularly at transitional zones of mucosal weakness (lesser curvature junction of antral and oxyntic mucosa), gastric ulcers form
- Progressive atrophy → intestinal metaplasia → dysplasia → gastric adenocarcinoma (the Correa cascade)
This explains the inverse relationship between DU (associated with high acid/antral pattern) and gastric cancer (associated with atrophy/pangastritis pattern) - both are caused by H. pylori but are mutually somewhat exclusive.
6. Carcinogenesis - The Correa Cascade
H. pylori is a WHO Group I carcinogen (definite human carcinogen). The proposed sequence for intestinal-type gastric adenocarcinoma is:
Normal mucosa → Chronic active gastritis → Atrophic gastritis → Intestinal metaplasia → Dysplasia → Adenocarcinoma
Key contributing factors:
-
CagA activates growth factor signaling (SHP-2, Ras-ERK, PI3K) mimicking sustained growth factor stimulation
-
Chronic inflammation produces mutagenic reactive oxygen/nitrogen species causing DNA strand breaks
-
Hypochlorhydria allows overgrowth of other bacteria that produce carcinogenic N-nitroso compounds
-
Sustained hypergastrinemia is trophic for gastric epithelium
-
H. pylori also causes polyclonal B-cell proliferation that can acquire driver mutations → gastric B-cell MALToma (MALT lymphoma) (eradication of H. pylori can cause MALToma regression)
-
Robbins, Cotran & Kumar Pathologic Basis of Disease; Yamada's Textbook of Gastroenterology, 7th ed.
7. Host Genetic Factors Modifying Disease
Not everyone with the same virulent strain develops the same disease. Host polymorphisms are key:
- IL-1β gene polymorphisms (particularly IL-1β -511 and +31): IL-1β is both a pro-inflammatory cytokine and a potent acid-suppressant. Polymorphisms causing ↑ IL-1β production → more atrophy/cancer risk rather than ulcer
- IL-1RA, TNF-α, IL-10 polymorphisms: modify mucosal inflammatory intensity
- Blood group O: increases H. pylori adhesion (H. pylori expresses adhesins that bind blood group antigens) → higher risk of PUD; blood groups A/B/AB have relative risk 0.75-0.91
- HLA-DQA1 genotype: associated with H. pylori-negative ulcer disease
Summary Table
| Feature | Antral gastritis | Pangastritis |
|---|---|---|
| Acid secretion | Increased | Normal/decreased |
| Gastrin | Elevated | Elevated |
| Somatostatin | Decreased | Decreased |
| Outcome | Duodenal ulcer | Gastric ulcer / adenocarcinoma |
| CagA role | Major (ulcer risk) | Major (cancer risk) |
Sources:
- Yamada's Textbook of Gastroenterology, 7th ed. - Chapters 49 & 10 (Pathogenesis of H. pylori-induced PUD and virulence factors)
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Chapter 17 (Pathogenesis section)
- Harrison's Principles of Internal Medicine, 22nd ed. - Chapter 335
- Medical Microbiology, 9th ed. - Chapter 28
- Mulholland and Greenfield's Surgery, 7th ed. - Helicobacter pylori virulence factors
This is a shared conversation. Sign in to Orris to start your own chat.