Pathophysiology, sequelae and management of renal tuberculosis.
Reading File
Finding Sources
Finding Sources
Searching PubMed
"renal tuberculosis" AND management
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
I now have comprehensive material from multiple authoritative sources. Let me compile the full answer.
Renal Tuberculosis: Pathophysiology, Sequelae, and Management
1. Pathophysiology
Route of Infection
Renal tuberculosis (TB) is part of genitourinary tuberculosis (GUTB), the most common form of extrapulmonary TB. The kidney is always infected hematogenously from a primary pulmonary focus. During the bacillaemic phase of primary infection, Mycobacterium tuberculosis seeds both kidneys bilaterally via the renal cortical capillaries. Bacilli lodge preferentially in the highly vascular glomerular tufts, where local oxygen tension and blood flow are high.
"Tuberculosis of the genitourinary tract is always secondary to tuberculosis elsewhere in the body. The primary site is usually the lungs." - Smith and Tanagho's General Urology, 19th Ed.
Latency and Reactivation
Initial seeding is typically subclinical. Cell-mediated immunity contains the organisms within granulomas. The lesions may remain dormant for years to decades before reactivating - hence many patients have no active pulmonary disease at the time of presentation (active TB elsewhere is found in fewer than half of patients with GUTB). Reactivation is triggered by immunosuppression, malnutrition, HIV co-infection, diabetes mellitus, or aging.
Progression Within the Kidney
The sequence of pathological events follows a predictable course:
| Stage | Pathological Change |
|---|---|
| Early | Microscopic granulomas in the cortex (glomerular tufts); cell-mediated response contains organisms |
| Sub-clinical | Granulomas enlarge; central caseous necrosis develops |
| Cavitation | Caseating granulomas rupture into the collecting system (calyces/pelvis), seeding the entire urinary tract downstream |
| Ulceration | Calyceal ulceration, "moth-eaten" calyces on imaging; papillary necrosis |
| Advanced | Parenchymal cavities, fibrosis, dystrophic calcification; eventually complete parenchymal replacement by caseous material or fibrous tissue |
| End-stage | "Cement kidney" or "putty kidney" - shrunken, functionless, fully calcified kidney (autonephrectomy) |
Microscopically: caseating granulomas with epithelioid cells, Langhans giant cells, lymphocytes, and plasma cells surround areas of amorphous caseous necrosis. Acid-fast staining demonstrates organisms. Progressive fibrosis destroys normal tubular architecture.
"In the most advanced stage of renal tuberculosis, the parenchyma may be completely replaced by caseous substance or fibrous tissue." - Smith & Tanagho, p. 245
Downstream Seeding
Once bacilli enter the collecting system, urine carries them to the ureter, bladder, and male genital tract (via the posterior urethra), establishing multi-focal disease. The cortex-to-medulla-to-pelvis-to-ureter-to-bladder sequence explains why:
- Bladder symptoms often dominate the clinical picture (frequency, dysuria)
- Only 75% of cases show unilateral disease on imaging
- Male genital involvement (prostate, seminal vesicles, epididymis) occurs via reflux or direct extension
2. Clinical Presentation
There is no classic clinical picture. Symptoms are predominantly vesical rather than renal in origin.
Constitutional symptoms:
- Low-grade fever, night sweats
- Malaise, fatiguability, weight loss
Urinary symptoms:
- Irritative voiding: frequency, urgency, dysuria, nocturia (from tuberculous cystitis)
- Gross or microscopic hematuria (renal or vesical origin)
- Flank dull ache (occasionally)
- Passage of clots, debris, or secondary calculi causing ureteral colic
Key clinical clues (Smith & Tanagho):
- Chronic cystitis failing to respond to standard antibiotic therapy
- Sterile pyuria - pyuria with a negative routine culture ("sterile pyuria" is the hallmark)
- Gross or microscopic hematuria
- Beaded/thickened vas deferens or nontender epididymal swelling
- Chronic scrotal draining sinus
- Prostatic nodularity / seminal vesicle thickening in a young man
3. Investigations
Urine Analysis
- Urinalysis is abnormal in >90% of patients with GUTB
- Sterile pyuria with hematuria is most common (acid urine - important contrast with alkaline urine of other infections)
- Routine urine culture grows no organisms ("sterile pyuria" prompts TB culture)
Urine Cultures
- Three consecutive early-morning urine specimens for mycobacterial culture (gold standard for diagnosis)
- Sensitivity: 75-90% for positive culture results
- Acid-fast smear of urine is usually positive, but culture confirmation is essential for drug susceptibility testing
- PCR / nucleic acid amplification testing (NAAT) of urine is more sensitive than culture and provides a faster diagnosis; culture is still needed for susceptibility testing
Tuberculin Skin Test / IGRAs
- TST positive in 60-90% of cases
- T-SPOT.TB (ELISPOT for IFN-γ) has pooled sensitivity ~91%; preferred over TST in BCG-vaccinated patients
- Third-generation QuantiFERON-TB Gold (QFT): sensitivity ~83%
Imaging
Plain KUB Radiograph:
- Calcifications present in >50% of patients
- Initial: faint punctate parenchymal calcifications
- Progressive: globular calcifications (tuberculoma)
- Papillary necrosis: triangular ring-like calcifications
- End-stage: pathognomonic lobar pattern - "putty/cement kidney" (calcific rims outlining individual renal lobes)
- Ureteral calcifications are characteristically intraluminal (vs. mural calcifications in schistosomiasis)
Intravenous Urography (IVU):
- Still considered gold standard for early disease detection
- Early: loss of sharpness/edge irregularities (uroepithelial erosion), "moth-eaten" calyces
- Progressive: papillary necrosis, cavity formation communicating with collecting system
- Infundibular stenosis with contrast excretion failure = "phantom calyx"
- Ureteral irregularity, multiple strictures, "pipe-stem" ureter
CT Urography (now preferred over IVP):
- Most common finding: renal calcification in 50%
- Identifies unilateral disease in 75% of cases
- Early: calyceal erosions, papillary necrosis
- Advanced: cavitation, cortical loss, uroepithelial thickening, dystrophic calcification, hydronephrosis
- Better characterizes perinephric extension, ureteral disease, and bladder involvement than IVU
Fig. 36.1 from Brenner & Rector's The Kidney - IVP and retrograde pyelogram showing unilateral hydronephrosis with distal ureteric narrowing:
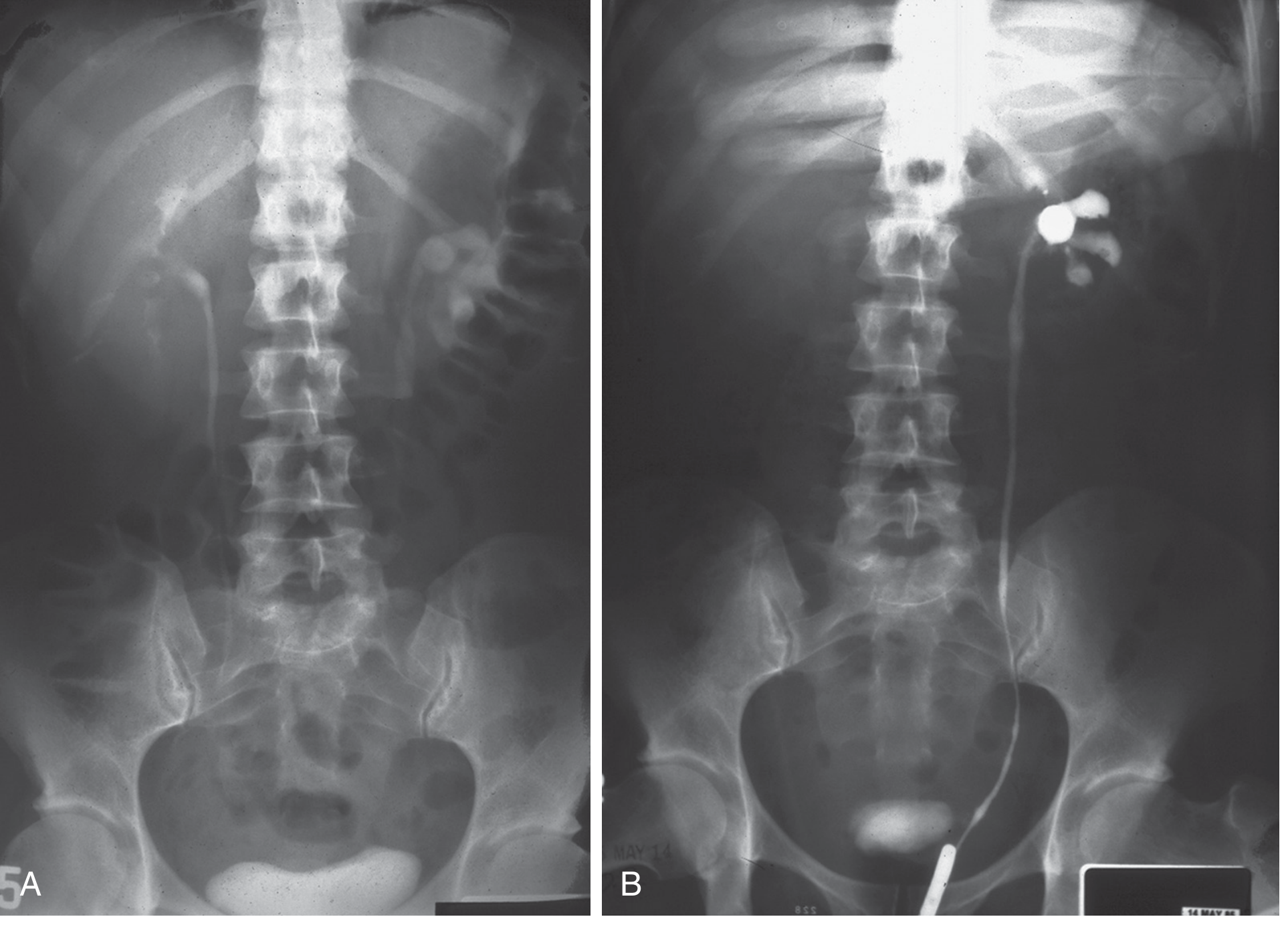
Cystoscopy:
- Early: mucosal hyperemia, white/yellow raised tubercles surrounded by halo of hyperemia
- Later: ulceration, reduced bladder capacity, scarring
- Reveals "golf-hole" ureteral orifices (gaping, rigid orifices) in advanced disease
Chest X-Ray
All patients should have a chest X-ray to exclude concomitant pulmonary TB. Chest radiograph is abnormal in 67-75% of patients.
Tissue Biopsy
- If suggestive renal abnormality with negative urine cultures: tissue biopsy confirms diagnosis
4. Sequelae
Renal Sequelae
| Sequela | Mechanism |
|---|---|
| Renal parenchymal cavitation | Caseation rupture into collecting system |
| Papillary necrosis | Ischemia + granulomatous destruction of papillae |
| Nephrocalcinosis / renal calculi | Dystrophic calcification; secondary stone formation in deformed pelvis (10% of patients) |
| Hydronephrosis | Infundibular or ureteropelvic stenosis from fibrosis |
| Perinephric abscess | Rare extension of caseation through renal capsule |
| Autonephrectomy | Complete calyceal/ureteral obstruction -> fibrotic, functionless "putty kidney" |
| Uremia | End-stage bilateral renal involvement |
Ureteral Sequelae
- Ureteral strictures are a characteristic lesion - most commonly at the juxtavesical (distal) third
- Multiple strictures along the ureter can create a "beaded" or "pipe-stem" appearance
- Progressive stricturing leads to hydronephrosis and eventual loss of renal function
- Complete obstruction causes autonephrectomy (the bladder urine may then appear normal, masking the problem)
Vesical Sequelae
- Tuberculous cystitis with mucosal ulceration
- Progressive fibrosis of the detrusor muscle -> contracted bladder (severely reduced capacity)
- Contracted bladder causes extreme urinary frequency (can be the dominant presenting complaint)
- Incompetence or stenosis of the ureterovesical junction -> vesicoureteric reflux -> hydronephrotic atrophy of the contralateral (uninvolved) kidney
- Late: irregular, calcified, small bladder
Genital Sequelae (Male)
- Epididymitis (classically nontender, hard swelling) with beaded vas deferens
- Bilateral epididymal obstruction -> sterility
- Epididymal abscess with scrotal skin involvement / chronic draining sinus
- Prostatic nodularity; seminal vesicle involvement
Systemic Sequelae
- Bilateral renal TB can lead to chronic kidney disease (CKD) and uremia
- Paradoxical reactions during treatment (inflammatory flares causing worsening of existing lesions)
5. Management
General Principles
GUTB is extrapulmonary TB. Medical therapy is primary; surgery is adjunctive and reserved for specific indications. Because bacilli exist in different micro-environments with varying metabolic rates, multiple drugs are essential to cover rapidly replicating, slowly replicating, and metabolically dormant populations, and to prevent emergence of drug resistance.
Before starting treatment: baseline CBC, liver function tests, renal function tests, HIV testing, ± hepatitis B/C and diabetes screening.
Medical Therapy - First-Line Regimen
Standard 6-Month Regimen (identical to pulmonary TB):
| Phase | Duration | Drugs | Notes |
|---|---|---|---|
| Intensive | 2 months | HRZE (Isoniazid + Rifampin + Pyrazinamide + Ethambutol) | Daily; all four drugs |
| Continuation | 4 months | HR (Isoniazid + Rifampin) | If isolate susceptible to first-line therapy |
"The treatment of genitourinary tuberculosis is similar to that of extrapulmonary tuberculosis at other sites. The initial regimen consists of four drugs (isoniazid, rifampin, pyrazinamide, and ethambutol) for 2 months, followed by two drugs (isoniazid and rifampin) for 4 months." - Brenner & Rector's The Kidney
Directly Observed Treatment (DOT) should be employed to ensure medication adherence and prevent development of drug-resistant strains.
Some clinicians extend treatment to 9 months (2 months HRZE + 7 months HR) for GUTB given the potential for scarring sequelae, though the WHO standard 6-month regimen is acceptable in drug-susceptible disease.
Second-Line Agents (MDR-TB or intolerance to first-line agents)
| Drug | Adult Dose | Key Adverse Effects |
|---|---|---|
| Streptomycin | 15 mg/kg IM/IV (max 1 g) | Vestibular/auditory toxicity, nephrotoxicity |
| Capreomycin | 15 mg/kg IM/IV (max 1 g) | Ototoxicity, renal damage, electrolyte imbalance |
| Amikacin/Kanamycin | 15 mg/kg IM/IV | Ototoxicity, nephrotoxicity |
| Cycloserine | 10-15 mg/kg PO in 2 doses | Psychiatric symptoms, seizures |
| Ethionamide | 15-20 mg/kg PO | GI toxicity, hepatotoxicity, optic neuritis |
| Levofloxacin/Moxifloxacin | Standard dosing | QT prolongation, GI |
| Linezolid | Standard dosing | Myelosuppression, peripheral neuropathy |
| Bedaquiline | Standard dosing | First new TB-specific drug in 40 years; QT prolongation |
Supportive Medical Measures
- Nutritional optimization - important for immune reconstitution
- Anticholinergic medications (e.g., oxybutynin) for bladder irritability symptoms
- Corticosteroids: may be used adjunctively for severe ureteral stricturing or threatened contralateral kidney (reduces fibrotic response); evidence base is limited
Surgical Management
Surgery is adjunctive - it establishes diagnosis or manages complications. The era of routine nephrectomy has passed with effective chemotherapy.
| Indication | Procedure |
|---|---|
| Non-functioning, destroyed kidney (autonephrectomy or symptomatic) | Nephrectomy (laparoscopic preferred) |
| Ureteral stricture (early, amenable) | Ureteral dilatation (>50% cure rate) |
| Ureteral stricture (recurrent/severe) | Ureteroplasty, endoscopic balloon dilatation, or ureteral stenting |
| Perinephric abscess | Drainage + delayed nephrectomy |
| Contracted bladder causing refractory symptoms | Augmentation cystoplasty (ileal or colonic patch) |
| Contracted bladder with incompetent UVJ threatening contralateral kidney | Urinary diversion (e.g., ileal conduit) - ureteroneocystostomy not feasible in fibrotic bladder |
| Ureteral reimplantation needed | Only after bladder capacity is restored |
"Ureteral stricture... ureteral dilatations offer a better than 50% chance of cure. The severely involved bladder may cause incompetence of the ureterovesical junction on the uninvolved side. Ureteroneocystostomy cannot be done in such a bladder; some form of urinary diversion may be required." - Smith & Tanagho, p. 247
Follow-Up and Monitoring
- CT urography or IVU every 6 months during and after treatment to detect ureteral scarring (can develop or progress even after successful chemotherapy)
- Urine cultures to confirm bacterial clearance
- Renal function tests serially, especially with bilateral disease or solitary kidney
- Long-term monitoring for paradoxical worsening (inflammatory flares 2-3 months into treatment)
- Drug susceptibility testing on isolated organism - mandatory before continuation phase
Summary Table
| Domain | Key Points |
|---|---|
| Pathophysiology | Hematogenous seeding -> cortical granulomas -> caseation -> cavitation rupturing into collecting system -> downstream dissemination |
| Hallmark lab finding | Sterile pyuria in acid urine |
| Imaging gold standard | CT urography (IVU for early disease); characteristic calcifications on KUB |
| Diagnosis | 3× early morning urine mycobacterial cultures (75-90% sensitive); PCR/NAAT faster but culture needed for susceptibility |
| Major sequelae | Ureteral stricture, hydronephrosis, contracted bladder, autonephrectomy, VUR threatening contralateral kidney, sterility |
| Medical treatment | 2 months HRZE + 4 months HR; DOT; baseline and monitoring LFTs/RFTs |
| Surgery | Nephrectomy for destroyed kidney; dilatation/stenting for strictures; augmentation/diversion for contracted bladder |
Sources:
- Brenner and Rector's The Kidney, 2-Volume Set - Genitourinary Tuberculosis (block22, p. 1646)
- Smith and Tanagho's General Urology, 19th Edition - Chapter 15, Genitourinary Tuberculosis (pp. 243-248)
- Campbell-Walsh-Wein Urology - Chapter 60 (block32, pp. 1718-1722)
- PubMed PMID 40254599 - Laparoscopic vs. open nephrectomy for inflammatory renal conditions (meta-analysis, 2025)
Etiopathology of congenital cystic disease of the kidney Describe the presentations of such anomaly
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
I now have comprehensive, authoritative content from multiple textbooks to write a thorough answer. Let me compile the full response.
Congenital Cystic Disease of the Kidney: Etiopathology and Presentations
Classification
Congenital renal cystic diseases comprise a heterogeneous group of disorders. The most clinically relevant categories are:
| Category | Inheritance | Gene(s) | Key Feature |
|---|---|---|---|
| ADPKD (Adult Polycystic KD) | Autosomal dominant | PKD1 (chr 16p13.3), PKD2 (chr 4q21) | Bilateral cortical/medullary cysts; adult onset |
| ARPKD (Childhood Polycystic KD) | Autosomal recessive | PKHD1 (chr 6p21) | Collecting duct ectasia; neonatal/infantile |
| Nephronophthisis-MCKD complex | AR (NPHP); AD (MCKD) | NPHP1-13; UMOD/uromodulin | Corticomedullary cysts; CKD in youth |
| Multicystic Dysplastic Kidney | Sporadic (mostly) | Unknown | Unilateral; ureteric bud-metanephric failure |
| Medullary Sponge Kidney | Sporadic | Unknown | Terminal collecting duct ectasia; benign |
1. Autosomal Dominant Polycystic Kidney Disease (ADPKD)
Epidemiology
ADPKD is one of the most common monogenic disorders: prevalence 1 in 400 to 1 in 1000. It accounts for approximately 10% of all chronic kidney disease and is a leading cause of ESKD worldwide.
Genetics
- PKD1 (chromosome 16p13.3): encodes polycystin-1 (PC1) - accounts for ~78-85% of pedigrees; more severe disease
- PKD2 (chromosome 4q21): encodes polycystin-2 (PC2) - accounts for ~10-15%; slower progression
- Two newer loci: GANAB and DNAJB11 with an ADPKD-like phenotype
- Each child of an affected parent has a 50% chance of inheriting the abnormal gene
- ~10% of cases arise from apparent de novo mutations
Pathogenesis - The "Two-Hit" and Ciliary Hypothesis
Although all renal tubular cells carry a heterozygous germline mutation, cysts develop in only a fraction of tubules. This is explained by the "two-hit" model: a somatic second-hit mutation in the wild-type allele is required for cyst initiation in that individual cell.
The key downstream mechanism involves the primary cilium:
- PC1 and PC2 co-localize to the primary cilia of tubular epithelial cells, where they function as mechanosensors of urinary flow
- PC1 is a large receptor-like transmembrane protein; PC2 functions as a calcium-permeable cation channel (TRP channel family)
- PC1 and PC2 form heterodimers - mutation in either gene gives the same phenotype
- Loss of PC1/PC2 function below a critical threshold disrupts mechanosensing and calcium influx into tubular cells
- Downstream consequences:
- Aberrant activation of the mTOR pathway (promotes cell proliferation)
- Increased cAMP signaling (promotes fluid secretion into cysts)
- Loss of normal cell polarity (mitotic spindle misorientation)
- Increased proliferation + increased transepithelial fluid secretion → cyst formation and progressive enlargement
"Reduction of polycystin-1 function below a critical threshold produces defects in mechanosensing by tubular epithelial cells that perturb downstream signaling events involving calcium influx. This in turn leads to altered cell polarity, increased proliferation, and increased fluid secretion, promoting the formation of cysts that progressively enlarge over time." - Robbins & Kumar Basic Pathology
Pathology
- Cysts arise from any level of the nephron (glomeruli, proximal/distal tubules, collecting ducts) and therefore show variable epithelial lining
- Both kidneys become massively enlarged - up to 4 kg each (normal ~150 g)
- Cysts up to 3-4 cm in diameter; fluid may be clear, turbid, or hemorrhagic
- Progressively replace intervening parenchyma (ischemic atrophy)
- Liver cysts in ~1/3 of patients (polycystic liver disease)
Clinical Presentation
ADPKD is typically asymptomatic until the 4th decade. Presentations include:
Renal:
- Flank pain / heavy dragging sensation - most common presenting symptom (from cyst expansion)
- Acute severe pain - from intracystic hemorrhage or obstruction
- Gross hematuria - intermittent; from cyst rupture into collecting system
- Hypertension - develops in ~75% of patients; often early (even before renal impairment)
- Urinary tract infections / infected cysts - fever, localized pain, pyuria
- Palpable bilateral flank masses on examination
- Nephrolithiasis - secondary stones
- Progressive renal impairment → ESKD (typically in 5th-6th decade for PKD1; later for PKD2)
Extrarenal manifestations:
| Manifestation | Frequency |
|---|---|
| Hepatic cysts (polycystic liver disease) | ~30-80% |
| Intracranial (berry) aneurysms of Circle of Willis | 10-30% - risk of subarachnoid haemorrhage |
| Mitral valve prolapse / cardiac valvular abnormalities | 20-25% |
| Pancreatic cysts | ~10% |
| Seminal vesicle cysts | ~40% in males |
| Arachnoid cysts | Less common |
Cause of death in ADPKD:
- ~40% coronary/hypertensive heart disease
- ~25% infection
- ~15% ruptured berry aneurysm or hypertensive intracerebral haemorrhage
2. Autosomal Recessive Polycystic Kidney Disease (ARPKD)
Epidemiology
Prevalence ~1 in 20,000 live births; carrier frequency ~1 in 70. Affects males and females equally.
Genetics
- All cases caused by mutations in PKHD1 (chromosome 6p21.1-p12)
- One of the largest genes in the human genome: ~470 kb, 67 exons
- Encodes fibrocystin (polyductin) - a 460 kDa single-pass transmembrane protein with a large extracellular domain
- Two truncating mutations in PKHD1 = lethal perinatal phenotype
- Surviving patients carry at least one non-truncating (hypomorphic) allele
- Significant genotype-phenotype correlation; sibling pairs may have markedly discordant severity (modifier genes)
Pathogenesis
- Fibrocystin localizes to primary cilia of tubular epithelial cells (like polycystins - confirming that ADPKD and ARPKD share a "ciliopathy" mechanism)
- Fibrocystin is also expressed in ureteric bud branches, biliary ducts, and pancreatic ducts during embryogenesis - explaining the combined renal-hepatic phenotype
- Fibrocystin may form a complex with PC2 and the kinesin motor KIF3B - disruption of this complex causes collecting duct cystogenesis
- Collecting ducts are the primary affected nephron segment (vs. any nephron segment in ADPKD)
Pathology
- Symmetrical, bilateral renal enlargement (up to >20× normal; combined weight ~300 g vs. normal ~25 g in neonates)
- Fusiform dilation of collecting ducts (1-2 mm), affecting up to 100% of collecting ducts in severe cases
- Dilated channels run at right angles to the cortical surface - creating a sponge-like appearance on cross-section
- Glomeruli and proximal tubular elements appear normal early on
- Liver involvement in essentially all cases: congenital hepatic fibrosis (CHF) with portal bile duct proliferation; Caroli disease (saccular intrahepatic bile duct dilatation) in variable proportion
- Renal-hepatic involvement exists on a spectrum: severe renal + mild hepatic (perinatal form) vs. mild renal + severe hepatic (juvenile/adult form)
Clinical Presentation
Perinatal / Neonatal form (most common):
- Oligohydramnios in utero (from fetal oliguria) → Potter sequence: pulmonary hypoplasia, limb deformities, characteristic facies
- Massively enlarged kidneys causing dystocia
- Respiratory failure at birth (pulmonary hypoplasia) - major cause of neonatal death
- Bilateral palpable abdominal masses
- Hypertension
Infantile/Juvenile forms (survivors):
- Renal insufficiency of variable severity
- Portal hypertension from congenital hepatic fibrosis: splenomegaly, oesophageal varices
- Cholangitis from Caroli disease
- Growth retardation, anaemia
- Progression to ESKD in childhood; liver transplantation may be required
"ARPKD is generally characterized by relatively rapid, symmetric, bilateral renal enlargement in infants due to collecting duct cysts in association with CHF." - Brenner & Rector's The Kidney
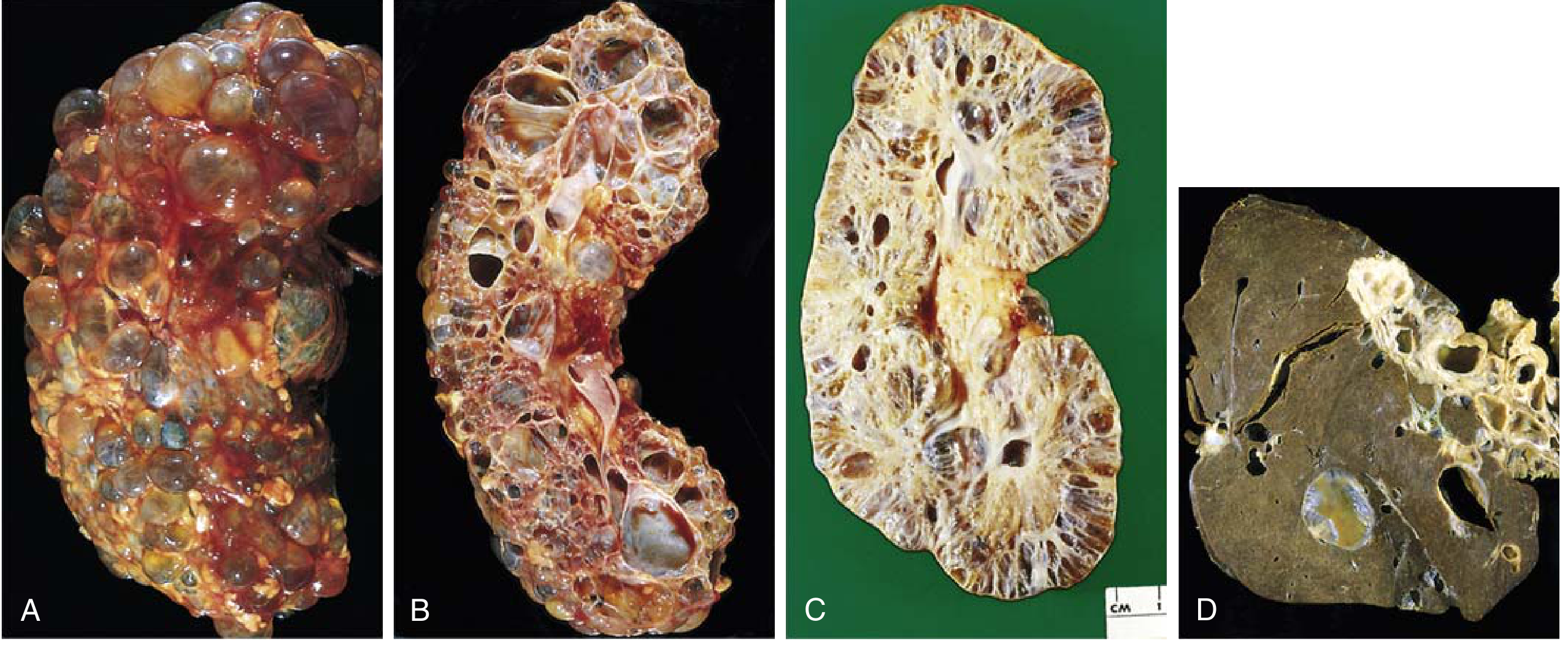
3. Nephronophthisis - Medullary Cystic Kidney Disease (NPHP-MCKD) Complex
Pathogenesis
- Nephronophthisis (NPHP): autosomal recessive; mutations in NPHP1-NPHP13 genes - all encoding ciliary/centrosomal proteins (nephrocystins)
- Medullary cystic kidney disease (MCKD): autosomal dominant; mutations in UMOD (uromodulin/Tamm-Horsfall protein gene) or MUC1
- Both share a ciliopathy mechanism with shared histopathology
- Cysts develop at the corticomedullary junction (rather than cortex or medulla)
- Additional histological features: tubular atrophy, thickened tubular basement membranes, chronic tubulointerstitial nephritis with progressive fibrosis
Morphology
- Small, contracted kidneys
- Cysts at the corticomedullary junction lined by flattened/cuboidal epithelium
- Chronic tubulointerstitial nephritis
Clinical Presentation
- Onset in childhood to young adulthood (juvenile form most common, ~15 yrs)
- Polyuria and polydipsia (earliest symptoms - tubular dysfunction, especially loss of urinary concentrating ability)
- Salt wasting (sodium-losing nephropathy)
- Anaemia disproportionate to degree of renal failure
- Progressive renal impairment → ESKD over 2-10 years
- Insidious; often no hypertension or proteinuria early
- Extrarenal manifestations in ~15-20% of NPHP: retinitis pigmentosa ± blindness (Senior-Løken syndrome), oculomotor apraxia, cerebellar malformations, hepatic fibrosis
- MCKD: associated with hyperuricemia, gout (UMOD mutations), and ESKD in adults
- Diagnosis is difficult: no serologic markers; cysts may be too small for imaging; requires sampling of corticomedullary junction on biopsy; genetic testing often needed
4. Multicystic Dysplastic Kidney (MCDK)
Etiopathology
- Most common form of renal cystic disease in childhood (~1:2400 to 1:4800 newborns)
- Results from abnormal ureteric bud-metanephric mesenchyme interaction during embryogenesis
- Failure of normal inductive signalling leads to:
- Abnormal nephrogenesis (no recognizable glomeruli)
- Obstructed or atretic ureter (possibly early complete ureteral obstruction raises hydrostatic pressure)
- Histological hallmarks: non-communicating cysts of variable size + dysplastic tubular epithelium surrounded by cuffs of cellular mesenchyme (primitive metanephric stroma)
Morphology
- Grossly distorted kidney replaced by non-communicating cysts (microscopic to several cm)
- No recognizable renal parenchyma; nonfunctional
- Usually unilateral (bilateral is lethal); contralateral kidney shows compensatory hypertrophy
Clinical Presentation
- Often detected on antenatal ultrasound (large cystic mass replacing kidney in fetus)
- Postnatal: palpable flank mass in neonate
- Usually asymptomatic if unilateral (contralateral kidney compensates)
- Complications: hypertension (rare), urinary tract infection, malignant transformation (very rare - Wilms tumour)
- Most undergo spontaneous involution by late childhood
- Bilateral MCDK: fatal (oligohydramnios, Potter sequence, pulmonary hypoplasia) - same consequence as severe ARPKD
5. Medullary Sponge Kidney (MSK)
Etiopathology
- Sporadic congenital disorder (no identified genetic mutation in most)
- Developmental abnormality of terminal collecting ducts - fusiform or cystic dilation
- May be related to overgrowth of ureteric bud branches during development
- Usually discovered incidentally in adults; benign condition
Pathology
- Dilation of terminal collecting ducts and small medullary cysts (2-7 mm)
- Kidney size is normal; medulla has a "sponge" or "paint-brush" appearance on IVU
- Predisposes to calcium oxalate/phosphate nephrolithiasis and infection within the ectatic ducts
Clinical Presentation
- Asymptomatic in most cases - discovered incidentally on imaging
- Symptomatic cases: renal colic (stone passage), haematuria, recurrent UTIs
- No progression to CKD in uncomplicated cases (unlike NPHP)
- Associated in rare cases with hemihypertrophy (Beckwith-Wiedemann syndrome)
Unifying Pathogenesis: The Ciliopathy Hypothesis
A major conceptual advance is the recognition that most hereditary cystic kidney diseases are ciliopathies - disorders of the primary (non-motile) cilium. PC1, PC2 (ADPKD), fibrocystin (ARPKD), nephrocystins (NPHP), and others all localize to the primary cilium or basal body. The cilium serves as a mechanosensor of urinary flow; disruption leads to:
- Loss of flow-induced Ca²⁺ signalling
- Dysregulation of mTOR and Wnt pathways
- Abnormal cell proliferation + fluid secretion
- Progressive cyst formation and expansion
This shared mechanism explains why different gene mutations cause overlapping phenotypes and why mTOR inhibitors (everolimus, sirolimus) and vasopressin V2 receptor antagonists (tolvaptan) have been explored therapeutically in ADPKD.
Summary Comparison Table
| Feature | ADPKD | ARPKD | NPHP-MCKD | MCDK | MSK |
|---|---|---|---|---|---|
| Inheritance | AD | AR | AR/AD | Sporadic | Sporadic |
| Gene | PKD1/PKD2 | PKHD1 | NPHP1-13 / UMOD | Unknown | Unknown |
| Protein | Polycystin-1/-2 | Fibrocystin | Nephrocystins | - | - |
| Cyst location | Any nephron segment | Collecting ducts | Corticomedullary junction | Whole kidney (dysplastic) | Terminal collecting ducts |
| Age at onset | 4th-5th decade | Perinatal/neonatal | Childhood/adolescence | Neonatal | Any age (usually adult) |
| Bilateral? | Yes | Yes | Yes | Usually unilateral | Yes |
| Key associations | Berry aneurysms, liver cysts | Congenital hepatic fibrosis, Caroli | Retinitis pigmentosa, gout (MCKD) | Contralateral UPJ obstruction | Nephrolithiasis |
| Outcome | ESKD in 50s-60s | ESKD in childhood; liver disease | ESKD in childhood/young adult | Involution; normal lifespan | Benign |
Sources:
- Robbins & Kumar Basic Pathology - Chapter 12, Kidney (pp. 524-526)
- Brenner and Rector's The Kidney, 2-Volume Set - Chapter 45, Hereditary Cystic Kidney Disorders (pp. 1983-2004)
- Campbell-Walsh-Wein Urology - Chapter 20, Kidney Development; Cystic Renal Disease section
- Comprehensive Clinical Nephrology, 7th Edition - Cystic kidney disease chapter
Principles of management of renal trauma
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive material from Sabiston, Campbell-Walsh-Wein, and Rosen's. Let me compile the full, authoritative answer.
Principles of Management of Renal Trauma
Overview
The kidney is the most commonly injured urinary tract organ. Approximately 80% of renal trauma is blunt (road traffic accidents, falls, contact sports) and 20% penetrating (stab wounds, gunshots). The fundamental goal of management is preservation of renal function while maintaining haemodynamic stability - a philosophy that has shifted dramatically from early surgical exploration to largely non-operative strategies.
"The prime goal of renal trauma management is preservation of renal function. Renal trauma has become largely nonoperative in modern times, especially in the setting of low- to intermediate-grade renal injuries." - Schwartz's Principles of Surgery
1. Initial Assessment
Primary Survey (ATLS)
Renal trauma rarely presents in isolation. The standard ATLS-based primary survey (Airway, Breathing, Circulation, Disability, Exposure) takes precedence. Haemodynamic status determines the entire subsequent management pathway.
Indicators of Renal Injury
History and mechanism:
- High-speed deceleration (MVA, fall from height) - risk of vascular pedicle injury and UPJ avulsion
- Direct flank impact, rib fractures, transverse process fractures
- Penetrating wounds to flank, back, lower chest, or abdomen
Examination clues:
- Flank pain/tenderness
- Flank ecchymosis ("Grey Turner's sign" in delayed cases)
- Visible haematuria
Haematuria
Urinalysis is the cardinal screening test. The first voided or catheterized specimen must be tested (later samples may be diluted by resuscitation fluids).
| Haematuria type | Significance |
|---|---|
| Gross haematuria | Always investigate with CT regardless of mechanism |
| Microscopic haematuria + haemodynamic shock (SBP <90 mmHg at any point) | Always investigate with CT |
| Microscopic haematuria alone, no shock | Usually no imaging required (<0.0016% significant injury) |
| Any haematuria in penetrating trauma | Always investigate |
| Rapid deceleration mechanism without haematuria | Still image - vascular pedicle injuries may present without haematuria |
"Although critical to the initial evaluation, the presence or absence of haematuria should not be the sole determinant. Mechanism of injury and concurrent injuries should also play a role." - Campbell-Walsh-Wein Urology
2. Classification: AAST Renal Organ Injury Scale (2018 Update)
The American Association for the Surgery of Trauma (AAST) Organ Injury Scale is the universally accepted classification and the key determinant of management decisions. The scale was updated in 2018 (Kozar et al.) to incorporate modern radiological and management data.
| Grade | Description | AIS |
|---|---|---|
| I | Subcapsular haematoma or parenchymal contusion without laceration | 2 |
| II | Perirenal haematoma confined to Gerota fascia; laceration ≤1 cm depth, no urinary extravasation | 2 |
| III | Laceration >1 cm depth without collecting system rupture or urinary extravasation; any vascular injury with active bleeding contained within Gerota fascia | 3 |
| IV | Parenchymal laceration extending into collecting system (urinary extravasation); renal pelvis laceration / UPJ disruption; active bleeding beyond Gerota fascia; segmental or complete infarction from vessel thrombosis (no active bleed); AVM or pseudoaneurysm | 4 |
| V | Main renal artery or vein laceration or avulsion of hilum; devascularized kidney with active bleeding; shattered kidney with loss of identifiable parenchymal anatomy | 5 |
Note: Advance one grade for bilateral injuries up to Grade III.
Grades I-III = low grade. Grades IV-V = high grade.
CT examples of renal trauma grades:
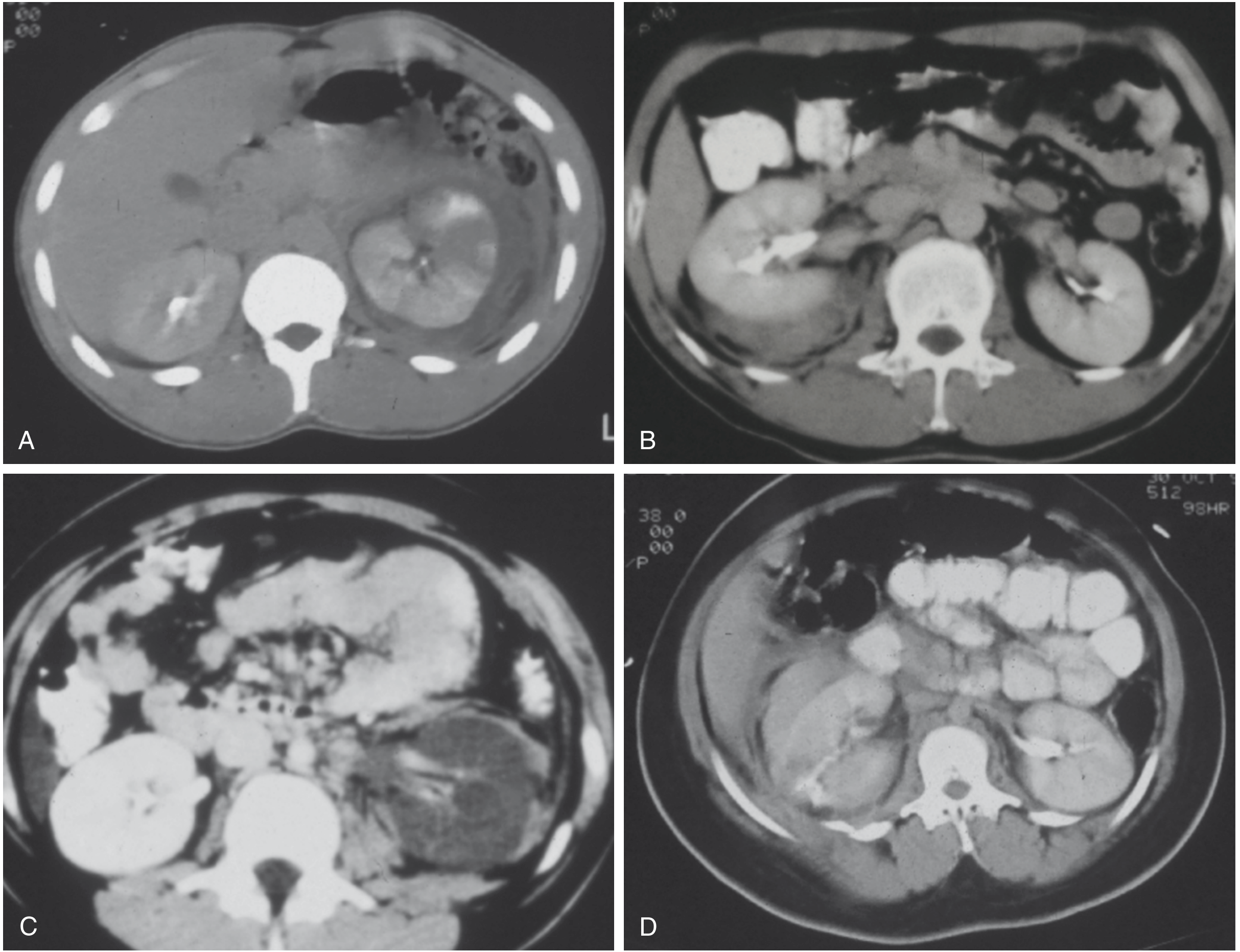
3. Imaging
Indications for Imaging (AUA / EAU Urotrauma Guidelines)
- All penetrating trauma with suspected renal involvement (flank, abdomen, ipsilateral rib fracture, flank ecchymosis, low chest wound) - if haemodynamically stable
- All blunt trauma with gross haematuria
- All blunt trauma with microhaematuria + hypotension (SBP <90 mmHg at any point)
- All blunt trauma with significant deceleration mechanism (high-speed MVA, fall from height) regardless of haematuria
- All paediatric patients with >5 RBCs/HPF
Imaging Modalities
CT with IV contrast - Gold Standard
- Contrast-enhanced CT abdomen/pelvis with immediate and delayed phase (at 9 minutes)
- Delayed images are essential to detect urinary extravasation (the 9-minute delay is optimal)
- If renal injury is seen on contrast phase, delayed imaging should be completed while the patient is still on the table
- CT findings predicting need for intervention:
- Perirenal haematoma >4 cm - every 1 cm increase = 66% higher likelihood of requiring intervention
- Intravascular contrast extravasation - threefold increase in odds of intervention
One-Shot Intravenous Pyelogram (IVP)
- Used when patient is too unstable for CT or requires immediate surgery
- 2 mL/kg IV contrast followed by a single abdominal radiograph
- Primarily used to confirm presence and function of the contralateral kidney before considering nephrectomy
- Has poor sensitivity and is difficult to perform well in hypoperfused patients; most trauma surgeons now use retroperitoneal palpation intraoperatively instead
FAST / Ultrasound
- FAST examination is part of the initial survey but has poor sensitivity and specificity for renal injuries in adults
- Cannot differentiate between haematoma and urine leak
- More useful in children; comparable diagnostic accuracy to CT in some paediatric series
- Can confirm bilateral kidney presence and detect retroperitoneal fluid collections
4. Management Decision Framework
The primary decision pivot is haemodynamic status:
Suspected Renal Trauma
│
▼
Haemodynamically UNSTABLE?
│
Yes ─┼──────────────────────────────► IMMEDIATE SURGICAL EXPLORATION
│ or Angioembolization if feasible
No │
▼
CT Imaging (with delayed phase)
│
├──► Grades I-III ─────────────► NON-OPERATIVE MANAGEMENT
│
├──► Grade IV ────────────────► Non-operative in most; surgical/
│ angioembolization if active bleed
│
└──► Grade V ─────────────────► Surgical or angioembolization;
selected cases: non-operative viable
5. Non-Operative Management (NOM)
NOM is now the standard of care for haemodynamically stable patients with AAST Grades I-IV renal injuries, regardless of mechanism (both blunt and selected penetrating).
Components of NOM
- Strict bed rest (historical; now fallen out of favour - ambulation with close monitoring is acceptable)
- Serial haematocrit monitoring
- Vital signs monitoring (haemodynamic stability)
- Analgesia, hydration, antibiotics (early antibiotics considered for all renal trauma patients to prevent UTI and perinephric abscess)
- Urinary catheterization to monitor output
Outcomes
- Grades I-III: nearly 100% success with NOM
- Grade IV: ~10% will eventually require open surgical procedure
- Grade V: ~50% require surgical intervention despite initial NOM attempt
- Paediatric renal salvage rate approaches 99% with modern NOM protocols
When to Obtain Repeat CT
- Not required routinely for all patients
- Indicated for: haemodynamic instability, falling haemoglobin, fever, worsening flank pain, abdominal distension
- All Grade IV-V injuries should have scheduled repeat CT to assess for urinoma, expanding haematoma, or abscess
Managing Urinary Extravasation on NOM
- Urinary extravasation in a haemodynamically stable kidney with intact vascularity can be managed conservatively - most heal spontaneously
- Large or expanding urinoma: treated with ureteral stent ± percutaneous drain
- Infected urinoma / perinephric abscess: percutaneous drainage + antibiotics
6. Angioembolization
Angioembolization is an increasingly important minimally invasive option that bridges NOM and surgery.
Indications
- Haemodynamically stable patients with CT evidence of:
- Active arterial contrast extravasation ("blush")
- Traumatic pseudoaneurysm
- Arteriovenous fistula
- High-grade injuries (IV-V) with ongoing haemorrhage not requiring immediate laparotomy
- Increasingly used for Grade IV and select Grade V injuries
Technique
- Superselective embolization - targets the bleeding segmental vessel, preserving as much viable parenchyma as possible
- Endovascular stents for acute renal artery thrombosis with intimal flap (requires anticoagulation)
- Can be combined with resuscitation
Outcomes
- Initial failure rate: 13-88% (variable across series)
- Subsequent re-embolization is highly successful
- Traumatic pseudoaneurysm: successful embolization in ~84.6% (Guyot et al.)
- Allows renal salvage that would otherwise result in nephrectomy
7. Surgical Management
Absolute Indications for Renal Exploration or Embolization
- Expanding, pulsatile retroperitoneal haematoma or renal pedicle avulsion
- Haemodynamic instability not responsive to resuscitation
- Suspected renal vascular pedicle avulsion
- Ureteropelvic junction avulsion
Relative Indications for Intervention
- Urinary extravasation in a devascularized renal unit
- Renal injury with concomitant colon or pancreatic injury
- Urinary extravasation from parenchymal injury (if not resolving)
- Persistent haemorrhage despite angioembolization
Operative Approach
- Transabdominal (midline) incision - allows complete visceral inspection and control of vessels before opening Gerota fascia
- Small bowel reflected cephalad; incision medial to inferior mesenteric vein extended to ligament of Treitz
- Early vascular control before opening Gerota fascia - limits blood loss and reduces nephrectomy rate
- Renal veins retracted cephalad to expose renal arteries (both run below renal veins)
- Colon reflected medially (left: white line of Toldt; right: hepatic attachments freed)
Renal Reconstruction (Renorrhaphy / Partial Nephrectomy)
When preservation is possible:
- Complete exposure of the kidney
- Temporary vascular control (vessel loops on renal artery/vein)
- Debridement of non-viable tissue
- Haemostasis (3-0 Vicryl suture bolstered with Surgicel/Nu-Knit)
- Closure of collecting system (watertight)
- Reapproximation of parenchymal defect; renorrhaphy
- Coverage with omental flap or Gerota fascia to promote healing and prevent fistula
- Closed suction drain placed at the repair site
Nephrectomy
- Performed when renal reconstruction is not feasible (shattered kidney, Grade V hilar avulsion, haemodynamic instability precluding time-consuming repair)
- Prior to nephrectomy: confirm presence and function of contralateral kidney (one-shot IVP or retroperitoneal palpation)
- Note: nephrectomy in the setting of high-grade renal trauma is associated with higher mortality than non-operative or reconstructive approaches - avoidance is preferred when possible
8. Complications
Early Complications
| Complication | Management |
|---|---|
| Persistent/recurrent haemorrhage | Angioembolization or re-exploration |
| Urinoma | Ureteral stent ± percutaneous drain |
| Perinephric abscess | Percutaneous drainage + antibiotics |
| Sepsis | IV antibiotics, source control |
| Urinary tract infection | Antibiotics (prophylactic early antibiotics for all renal trauma) |
Late Complications
| Complication | Notes |
|---|---|
| Renovascular hypertension | From renal artery stenosis/thrombosis post-trauma ("Page kidney" from external compression by haematoma); most commonly within 1 year; treat with nephrectomy or revascularisation |
| UPJ stricture | From healing of collecting system lacerations |
| Arteriovenous fistula | Presents with haematuria weeks later; treat with superselective embolization |
| Delayed pseudoaneurysm rupture | Haematuria weeks post-injury; embolization |
| Chronic renal failure | If bilateral injury or solitary kidney |
9. Special Considerations
Penetrating Renal Trauma
- Historically mandated urgent exploration; modern approach more selective
- Isolated penetrating renal injuries without abdominal involvement: observe or embolize
- Retroperitoneal violation with peritoneal contamination: usually requires surgical exploration
- Morbidity reduced if operative management is performed in a delayed rather than immediate fashion when patient allows
Paediatric Renal Trauma
- Kidneys more vulnerable in children (less perirenal fat, larger relative kidney size, weaker abdominal musculature)
- CT preferred; consider radiation dose (a single delayed phase may avoid subsequent imaging)
- NOM is even more strongly favoured - renal salvage rate approaches 99%
- Lower threshold for imaging: >5 RBCs/HPF warrants CT
Solitary Kidney / Pre-existing Renal Anomaly
- Higher stakes - every effort made for renal preservation
- Angioembolization strongly preferred over nephrectomy
Summary Algorithm
| Scenario | Management |
|---|---|
| Haemodynamically unstable | Immediate surgical exploration ± angioembolization |
| Grade I-III, stable | NOM: observation, serial haematocrit, monitoring |
| Grade IV, stable, no active bleed | NOM; repeat imaging; ureteral stent if urinoma |
| Grade IV, stable, active arterial bleed | Angioembolization |
| Grade V, selected, stable | NOM or angioembolization; 50% will need surgery |
| Grade V, unstable / hilar avulsion | Surgical exploration; renorrhaphy or nephrectomy |
| Expanding pulsatile haematoma | Immediate exploration regardless of grade |
| Urinary extravasation, stable vasculature | Ureteral stent ± percutaneous drain |
Sources:
- Sabiston Textbook of Surgery, 21st Edition - Chapter 39, Renal Trauma (pp. 748-749)
- Campbell-Walsh-Wein Urology - Chapter 90, Renal Trauma (pp. 2643-2648)
- Rosen's Emergency Medicine - Chapter 39, Urinary Tract Trauma
- Schwartz's Principles of Surgery, 11th Edition - Kidneys
- AAST Organ Injury Scaling 2018 Update: Kozar RA et al., J Trauma Acute Care Surg. 2018;85:1119-1122
Causes of unilateral hydronephrosis
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive, well-organized content from Harrison's, Brenner & Rector's, Robbins, and Bailey & Love's. Let me now compile the full, structured answer.
Causes of Unilateral Hydronephrosis
Key principle: When obstruction occurs above the level of the bladder (i.e., at or proximal to the ureterovesical junction), dilatation is unilateral. Lesions at or below the bladder tend to cause bilateral hydronephrosis. Unilateral hydronephrosis can also result from vesicoureteral reflux (non-obstructive).
Causes are best organized by location (intrarenal → UPJ → ureter → UVJ) and mechanism (intrinsic intraluminal, intrinsic intramural, extrinsic, functional).
I. Congenital / Developmental Causes
A. Ureteropelvic Junction (UPJ) Obstruction
- The most common cause of unilateral hydronephrosis overall - incidence ~1 in 500 live births
- More common in males; more commonly affects the left side; bilateral in ~10%
- Intrinsic (most common): Aperistaltic segment at the UPJ due to muscular hypoplasia / deficient smooth muscle investment - urine cannot be propagated across the segment
- Intrinsic anatomical: High insertion of the ureter into the renal pelvis; persistent mucosal folds; ureteral polyp
- Extrinsic: Crossing aberrant lower pole vessel (most common extrinsic cause) compressing the UPJ; fibrous bands
B. Ureterovesical Junction (UVJ) Obstruction / Obstructed Megaureter
- Intrinsic aperistaltic segment at the UVJ prevents drainage
- Results in hydroureteronephrosis (dilated ureter + renal pelvis)
C. Ureterocele
- Cystic dilatation of the intravesical portion of the ureter
- Obstructs the ureteral orifice; usually associated with a duplex collecting system (upper moiety ureter)
- May cause ipsilateral hydronephrosis ± contralateral obstruction
D. Retrocaval (Circumcaval) Ureter
- Rare anomaly: right ureter loops behind and around the inferior vena cava
- Causes right-sided ureteral obstruction with characteristic "reverse J" or "fishhook" deformity on IVU/IVP
- Extrinsic compression by the IVC on the ureter
E. Vesicoureteral Reflux (VUR)
- Abnormal insertion of the ureter into the bladder (short submucosal tunnel)
- Not strictly obstructive - reflux of urine from bladder to kidney causes intermittent hydronephrosis
- Can be unilateral; may resolve spontaneously with age
- Risk: recurrent pyelonephritis and renal scarring
F. Ureteral Duplication with Incomplete Drainage
- Duplex kidney with obstructed upper moiety
- Upper pole ureter tends to have ectopic insertion (below normal orifice) and is prone to obstruction/ureterocele
II. Acquired Intrinsic Causes
Intraluminal (within the ureteral lumen)
| Cause | Details |
|---|---|
| Urolithiasis (calculi) | Most common acquired cause in adults; stones typically lodge at three sites: UPJ, pelvic brim (iliac vessels), UVJ; causes acute unilateral obstructive uropathy and renal colic |
| Blood clots | From renal trauma, tumour haemorrhage, or arteriovenous malformations; clot occludes the ureter |
| Sloughed renal papillae | In analgesic nephropathy, diabetic nephropathy, sickle cell disease, TB; detached papilla obstructs ureter |
| Fungus ball | Candida albicans/tropicalis - intraluminal fungal mass; more common in immunocompromised patients |
| Pyonephrosis debris | Purulent material obstructing a chronically infected collecting system |
Intramural (within the ureteral wall)
| Cause | Details |
|---|---|
| Ureteral stricture - iatrogenic | Post-surgical (hysterectomy, colonic resection, ureteroscopy, pelvic surgery); radiation-induced stricture (after cervical, colorectal cancer treatment) |
| Ureteral stricture - inflammatory | Tuberculosis (most commonly affects distal third of ureter; multiple strictures); Schistosomiasis (Schistosoma haematobium - chronic bilharzial fibrosis) |
| Ureteral stricture - trauma | External trauma, ureteral instrumentation |
| Primary ureteral tumour | Transitional cell carcinoma (urothelial carcinoma) of the ureter - uncommon; causes progressive obstruction |
| Ureteral polyp | Benign fibrous/inflammatory polyp |
III. Acquired Extrinsic Causes
Extrinsic compression of the ureter by structures outside it - divided by anatomical region:
At the Renal Pelvis / UPJ Level
- Crossing lower-pole vessel (may also be developmental - see above)
- Perinephric haematoma (post-trauma or spontaneous) - external compression of pelvis
- Urinoma - compresses collecting system
Along the Course of the Ureter
Vascular:
- Iliac artery aneurysm (abdominal aortic or iliac) - extrinsic compression
- Ovarian vein thrombosis / ovarian vein syndrome - right-sided unilateral obstruction (right ovarian vein runs parallel to right ureter)
- Lymphocele after pelvic lymph node dissection
Gynaecological:
- Carcinoma of the cervix - most common gynaecological cause; direct invasion or parametrial extension compresses ureter
- Carcinoma of the ovary / uterus - direct extension into the retroperitoneum
- Endometriosis - pelvic endometriosis involves the ureter (extrinsic compression or intrinsic invasion); onset insidious; often affects left ureter; obstruction may be cyclical
- Pregnancy - physiological hydronephrosis of pregnancy (right > left; due to the gravid uterus and dextrorotation); usually asymptomatic; more pronounced on the right
Gastrointestinal / Colorectal:
- Carcinoma of the colon / sigmoid - particularly sigmoid or caecal tumours invading the retroperitoneum
- Crohn's disease - retroperitoneal fistula or inflammatory mass can involve the right ureter
- Appendix abscess - can compress the right ureter
- Diverticular disease - pericolic abscess/fistula involving left ureter
Retroperitoneal Pathology:
- Retroperitoneal fibrosis (RPF) - idiopathic (Ormond's disease) or secondary (drugs: methysergide, pergolide; inflammation; aortic aneurysm; malignancy; TB; actinomycosis; sarcoidosis; iatrogenic); thick fibrous plaque encases the ureters; characteristically pulls them medially; usually bilateral but may be asymmetric / unilateral
- Retroperitoneal lymphadenopathy - lymphoma (especially follicular lymphoma); metastatic cancer (testicular, prostate, cervical, colonic); causes extrinsic ureteral compression
- Retroperitoneal tumours - sarcoma, liposarcoma, other primary retroperitoneal tumours
- Retroperitoneal haematoma - post-traumatic or from aortic aneurysm rupture
- Psoas abscess - can involve adjacent ureter
- Enlarged prostate / prostate cancer - at the UVJ level; strictly a bilateral cause at the bladder outlet but can present asymmetrically
Iatrogenic / Surgical:
- Accidental ureteral ligation during hysterectomy, colonic resection, or aortic surgery - causes acute unilateral hydronephrosis; may go undetected if unilateral
- Ureteral injury from pelvic surgery - unilateral obstruction
IV. Functional / Non-Mechanical Causes
| Cause | Mechanism |
|---|---|
| Vesicoureteral reflux | Non-obstructive; urine refluxes back up from bladder; dilates renal pelvis without true obstruction |
| Prune belly syndrome (Eagle-Barrett) | Absent abdominal musculature + megaureters + cryptorchidism; poor ureteral peristalsis |
| Primary megaureter (functional) | Aperistaltic distal ureteral segment; urine not propelled efficiently; may be unilateral |
| Neurogenic bladder | High-pressure bladder transmits pressure retrograde; usually bilateral but can be asymmetric |
V. Renal Pelvis / Collecting System Tumours
- Transitional cell carcinoma (urothelial carcinoma) of the renal pelvis - intrinsic obstruction from tumour mass in the pelvis/infundibulum
- Renal cell carcinoma - large tumour invading/obstructing the collecting system
- Wilms' tumour (nephroblastoma) - in children; large mass distorting pelvicalyceal system
VI. Inflammatory / Infective Causes
| Cause | Mechanism |
|---|---|
| Renal tuberculosis | Ureteral strictures (especially juxtavesical); may also cause infundibular stenosis ("phantom calyx") |
| Schistosomiasis (bilharziasis) | Chronic ureteral fibrosis; intramural calcification; distal ureteral obstruction |
| Ureteritis (non-specific) | Mucosal oedema; usually temporary |
| Pyelonephritis / perinephric abscess | Adjacent inflammatory mass compressing pelvis |
| Xanthogranulomatous pyelonephritis | Inflammatory mass replacing renal parenchyma; can obstruct collecting system |
Summary Classification
UNILATERAL HYDRONEPHROSIS
│
├── CONGENITAL
│ ├── UPJ obstruction (intrinsic aperistaltic / crossing vessel)
│ ├── UVJ obstruction / obstructed megaureter
│ ├── Ureterocele
│ ├── Retrocaval ureter (right-sided)
│ └── VUR (reflux nephropathy)
│
├── ACQUIRED - INTRINSIC
│ ├── Intraluminal: Calculus, blood clot, sloughed papilla, fungus ball
│ └── Intramural: Stricture (TB, schistosomiasis, iatrogenic, radiation), TCC ureter
│
├── ACQUIRED - EXTRINSIC
│ ├── Gynaecological: Ca cervix, Ca ovary, endometriosis, pregnancy
│ ├── GI/colorectal: Ca colon, Crohn's disease, diverticular abscess
│ ├── Vascular: Aortic/iliac aneurysm, ovarian vein syndrome
│ ├── Retroperitoneal: RPF, lymphoma, retroperitoneal sarcoma, haematoma
│ ├── Iatrogenic: Ureteral ligation during surgery
│ └── Renal mass: RCC, TCC renal pelvis, Wilms'
│
└── FUNCTIONAL
└── Reflux, neurogenic bladder (asymmetric), primary megaureter
"Obstruction to urine flow can result from intrinsic or extrinsic mechanical blockade as well as from functional defects. When lower urinary tract obstruction is above the level of the bladder, unilateral dilatation of the ureter (hydroureter) and renal pyelocalyceal system (hydronephrosis) occurs; lesions at or below the level of the bladder cause bilateral involvement." - Harrison's Principles of Internal Medicine, 22nd Edition
Sources:
- Harrison's Principles of Internal Medicine, 22nd Ed. - Chapter 331, Obstructive Uropathy (Table 331-1)
- Brenner & Rector's The Kidney, 2-Volume Set - Chapter 37, Acquired Causes of Obstruction
- Bailey & Love's Short Practice of Surgery, 28th Ed. - Chapter 82, PUJO and Hydronephrosis
- Robbins, Cotran & Kumar: Pathologic Basis of Disease - Chapter 21, Disorders of the Ureters
Types pf renal calculi
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive, authoritative content from multiple textbooks. Here is the full, detailed answer.
Types of Renal Calculi (Urolithiasis)
General Principles of Stone Formation
Kidney stones form when the urinary concentration of stone-forming constituents exceeds their solubility (supersaturation) in urine. An organic mucoprotein matrix (1-5% by weight) is present in all calculi and serves as a scaffold. Key factors influencing formation:
- Supersaturation of urine with stone-forming salts
- Reduced urine volume (dehydration, low fluid intake)
- Urinary pH - acidic urine favours uric acid and cystine stones; alkaline urine favours calcium phosphate and struvite stones
- Deficiency of crystallization inhibitors: citrate, pyrophosphate, magnesium, glycosaminoglycans, osteopontin, nephrocalcin
- Randall plaques - calcium phosphate deposits on renal papillae serving as nidus
- Stasis - promotes formation of smooth, round "milk of calcium" stones
"Increased concentration of stone constituents, changes in urinary pH, decreased urine volumes, and the presence of bacteria influence the formation of calculi." - Robbins, Cotran & Kumar
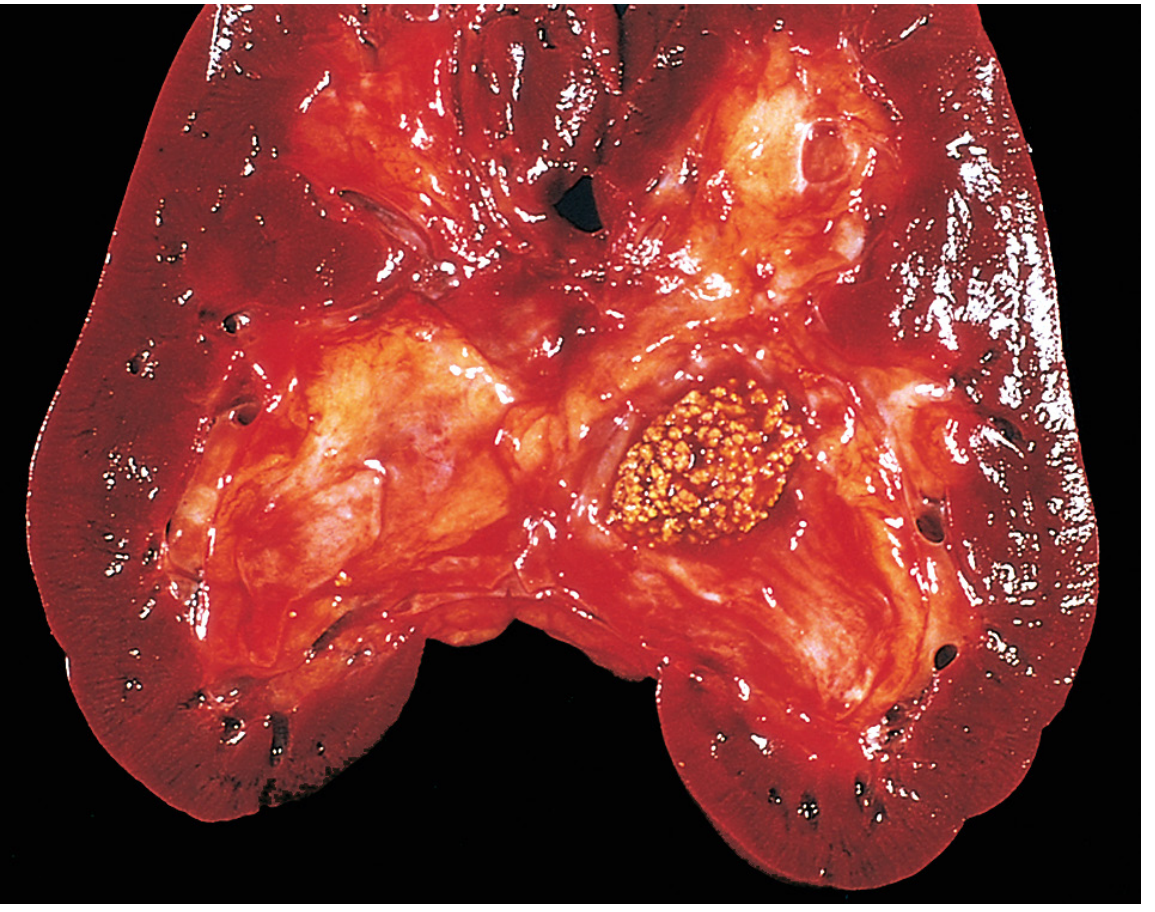
Prevalence Summary Table
| Stone Type | Composition | Frequency |
|---|---|---|
| Calcium oxalate / Calcium phosphate | Calcium salts | 70-80% |
| Uric acid | Monosodium urate | 5-10% |
| Struvite (Magnesium ammonium phosphate) | Triple phosphate | 5-10% |
| Cystine | Amino acid | 1-2% |
| Drug-induced / Rare | Various | <1% |
1. Calcium Stones (70-80%)
The most common type. Subdivided into:
A. Calcium Oxalate Stones (~60-65% of all stones)
- Most common renal stone overall
- Appear as dark brown/yellow, hard, spiculated "mulberry" stones on gross examination
- Radiopaque on plain X-ray
- Appear as envelope or dumbbell crystals on urine microscopy
Metabolic causes / Risk factors:
| Mechanism | Cause | Details |
|---|---|---|
| Hypercalciuria (most common - ~55%) | Absorptive type I | Increased intestinal Ca absorption; high Na/protein diet |
| Absorptive type II | Diet-dependent; corrects with low-Ca diet | |
| Renal hypercalciuria | Impaired tubular reabsorption of Ca | |
| Resorptive (hypercalcaemic) | Hyperparathyroidism (most common hypercalcaemic cause), sarcoidosis, diffuse bone disease, vitamin D toxicity, malignancy | |
| Hyperuricosuria (~20%) | Gout, high purine intake | Uric acid crystals nucleate calcium oxalate deposition in collecting ducts |
| Hyperoxaluria (~5%) | Primary hyperoxaluria (AR - defect in oxalate metabolism) | Very high urinary oxalate; can cause nephrocalcinosis |
| Enteric hyperoxaluria | IBD, short bowel syndrome - fat malabsorption causes Ca-fatty acid binding, leaving oxalate free to be absorbed | |
| Dietary hyperoxaluria | Excess spinach, nuts, chocolate; low dietary calcium intake | |
| Hypocitraturia | RTA type I, chronic diarrhoea, metabolic acidosis | Citrate is a key inhibitor; binds Ca in urine; reduced in acidosis |
| Idiopathic | No identifiable metabolic defect | ~15-20% of calcium stone formers |
B. Calcium Phosphate Stones
- Pure calcium phosphate stones are rare; usually mixed with calcium oxalate
- Common forms:
- Hydroxyapatite - associated with infection, alkaline urine, hyperparathyroidism
- Brushite (calcium hydrogen phosphate) - characteristically associated with distal renal tubular acidosis (type I RTA); alkaline urine required; may recur rapidly after lithotripsy
- Radiopaque; soft and chalky in consistency
- Alkaline urine (pH >6.5) promotes calcium phosphate crystallization
2. Uric Acid Stones (5-10%)
- Account for 10-15% of all stones
- Important feature: radiolucent on plain X-ray (KUB) - invisible; only seen on CT
- Hard, smooth, yellow-brown stones
- Uric acid is poorly soluble at urine pH <5.5; remains in solution at pH >6.5
Causes / Risk factors:
| Mechanism | Cause |
|---|---|
| Hyperuricosuria | Gout, myeloproliferative disorders (leukaemia, rapid cell turnover), tumour lysis syndrome, high purine/protein diet |
| Persistently acid urine (pH <5.5) | Chronic diarrhoea, metabolic syndrome, obesity, diabetes mellitus type 2, Lesch-Nyhan syndrome |
| Lesch-Nyhan syndrome | X-linked recessive deficiency of HGPRT; severe hyperuricaemia |
| Idiopathic | >50% of uric acid stone patients have neither hyperuricaemia nor hyperuricosuria; low urine pH is the primary driver |
Key: Uric acid stones are the only radiolucent stones commonly encountered - they are invisible on plain X-ray but clearly visible on non-contrast CT.
Treatment principle: Urinary alkalinization (sodium bicarbonate or potassium citrate to raise pH to 6.5-7.0) can dissolve existing uric acid stones - the only type amenable to medical dissolution.
3. Struvite Stones - "Infection Stones" (5-10%)
Also called triple phosphate or magnesium ammonium phosphate (MAP) stones.
- Composition: Mg NH₄ PO₄ · 6H₂O (struvite) ± calcium carbonate apatite
- Soft, pale yellow/white, chalky texture; grow rapidly and fill the entire pelvicalyceal system
- Radiopaque (though less dense than calcium stones)
Pathogenesis
The critical event is infection by urease-producing bacteria:
Urease
Urea ──────────► CO₂ + NH₃ (ammonia)
↓
Alkaline urine (pH >7)
↓
Supersaturation with Mg²⁺, NH₄⁺, PO₄³⁻
↓
Struvite crystal precipitation
Urease-producing organisms:
- Proteus mirabilis (most common)
- Klebsiella pneumoniae
- Staphylococcus aureus / epidermidis
- Serratia marcescens
- Enterobacter species
- Pseudomonas aeruginosa
- Providencia species
- Corynebacterium urealyticum
- Note: Escherichia coli does NOT produce urease and does NOT cause struvite stones
Staghorn Calculi
- Struvite stones are the most common cause of staghorn calculi - branching stones that take the shape of the entire pelvicalyceal system
- Can also be formed by cystine, uric acid, and calcium oxalate
- Grow rapidly; often asymptomatic until very large
- Associated with recurrent UTIs, pyonephrosis, and eventual renal destruction
- Complete clearance is mandatory - residual fragments cause rapid recurrence and persistent bacteriuria
- More common in females (due to higher UTI prevalence)
4. Cystine Stones (~1-2%)
- The rarest of the major stone types
- Pale yellow, waxy, smooth appearance; very hard due to disulphide bonds
- Faintly radiopaque (semi-opaque - ground glass appearance on plain X-ray)
- Hexagonal crystals on urine microscopy (pathognomonic)
Pathogenesis
- Caused by cystinuria - an autosomal recessive defect in the renal tubular and intestinal transport of dibasic amino acids: Cystine, Ornithine, Lysine, Arginine (mnemonic: COLA)
- Genetic defect in the SLC3A1 (type A) or SLC7A9 (type B) transporter genes
- Cystine is poorly soluble at normal and acidic urine pH; only becomes soluble at pH >7.5
- Forms in early life - often presents in children and young adults
Key points:
- Very hard - poorly amenable to shock wave lithotripsy (SWL); often requires ureteroscopy or PCNL
- Recurrent; lifelong management required (high fluid intake, alkalinization, D-penicillamine or tiopronin)
- Cyanide-nitroprusside test is positive (screening test for cystinuria)
5. Drug-Induced / Rare Stones (<1%)
| Drug/Cause | Stone Type | Notes |
|---|---|---|
| Indinavir (protease inhibitor for HIV) | Indinavir stone | Radiolucent; not visible on CT |
| Ritonavir | Similar | Radiolucent |
| Triamterene | Triamterene stone | Rare; diuretic metabolite |
| Sulfonamides / Sulfadiazine | Drug crystal | Intrarenal precipitation |
| Acyclovir | Crystal nephropathy | Large doses; IV acyclovir |
| Ciprofloxacin | Crystalluria | Especially in dehydrated patients |
| Adenine phosphoribosyltransferase (APRT) deficiency | 2,8-dihydroxyadenine | AR; mimics uric acid stones; radiolucent |
| Xanthinuria | Xanthine stones | Very rare; radiolucent |
Comparative Summary
| Feature | Calcium Oxalate | Calcium Phosphate | Uric Acid | Struvite | Cystine |
|---|---|---|---|---|---|
| Frequency | 60-65% | 10-15% (mixed) | 5-10% | 5-10% | 1-2% |
| Radiopacity | ✔ Radiopaque | ✔ Radiopaque | ✘ Radiolucent | ✔ Radiopaque | Faintly opaque |
| Urine pH | Acid | Alkaline (>6.5) | Acid (<5.5) | Alkaline (>7) | Acid |
| Crystal shape | Envelope/dumbbell | Coffin lid / amorphous | Rhombus/needle | Coffin lid | Hexagonal |
| Colour | Brown/yellow | White/chalky | Yellow-brown | Pale yellow | Yellow/waxy |
| Hardness | Hard | Soft/chalky | Hard | Soft | Very hard |
| Gender | M > F | - | M > F | F > M | Equal |
| Key association | Hypercalciuria, hyperoxaluria | RTA, infection | Gout, obesity | Proteus UTI | Cystinuria (AR) |
| SWL efficacy | Good | Good | Good | Moderate | Poor |
| Dissoluble? | No | No | Yes (alkalinize) | Partial (antibiotics) | Partial |
Inhibitors of Stone Formation (Clinically Relevant)
The following urinary substances inhibit crystallization and stone growth:
- Citrate - chelates calcium; most important inhibitor; low in RTA and metabolic acidosis
- Magnesium - competes with calcium to bind oxalate
- Pyrophosphate - inhibits calcium phosphate nucleation
- Glycosaminoglycans (chondroitin sulphate, heparan sulphate)
- Osteopontin - inhibits crystal aggregation and adhesion
- Nephrocalcin (Tamm-Horsfall protein) - inhibits calcium oxalate crystal growth
Deficiency of these inhibitors, especially hypocitraturia, is an independent risk factor for calcium stone formation even in the absence of supersaturation.
Sources:
- Robbins, Cotran & Kumar: Pathologic Basis of Disease - Chapter 20, Table 20.12 (pp. 877-878)
- Bailey & Love's Short Practice of Surgery, 28th Ed. - Chapter 82, Types of Stones (pp. 1495-1497)
- Sabiston Textbook of Surgery, 21st Ed. - Urolithiasis, Background
- Barash, Cullen & Stoelting's Clinical Anaesthesia, 9th Ed. - Table 50-9, Spectrum of Kidney Stone Types
What are the segments of kidney?
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Loading Image
Finding Sources
Reading File
I now have all the content needed. Here is a thorough, well-illustrated answer.
Segments of the Kidney
The kidney is divided into 5 arterial segments (Graves' segmentation, 1954) based on the distribution of the segmental arteries arising from the renal artery. This is purely a vascular/functional segmentation - each segment is supplied by a terminal (end) artery with no anastomoses between adjacent segments. Infarction of one segment does not affect another, but occlusion of a segmental artery causes irreversible ischaemia of its territory.
The Five Renal Segments (Graves, 1954)
| Segment | Abbreviation | Blood Supply | Surface Territory |
|---|---|---|---|
| Apical | A | Apical segmental artery (from anterior division, occasionally posterior) | Upper pole - both anterior and posterior surfaces |
| Upper (Anterosuperior) | U | Upper segmental artery (anterior division) | Anterior surface, upper third |
| Middle (Anteromedial) | M | Middle segmental artery (anterior division) | Anterior surface, middle third |
| Lower | L | Lower segmental artery (anterior division, shared with posterior) | Lower pole - both anterior and posterior surfaces |
| Posterior | P | Posterior segmental artery (posterior division of renal artery) | Entire posterior surface except apical and lower poles |
"The anterior half of the kidney can be divided into upper (U), middle (M), and lower (L) segments, each supplied by a segmental branch of the anterior division of the renal artery. A small apical segment (A) is usually supplied by a division from the anterior segmental branch. The posterior half of the kidney is divided into apical (A), posterior (P), and lower (L) segments, each supplied by branches of the posterior division of the renal artery."
- Brenner & Rector's The Kidney (modified from Graves FT, Br J Surg, 1954)
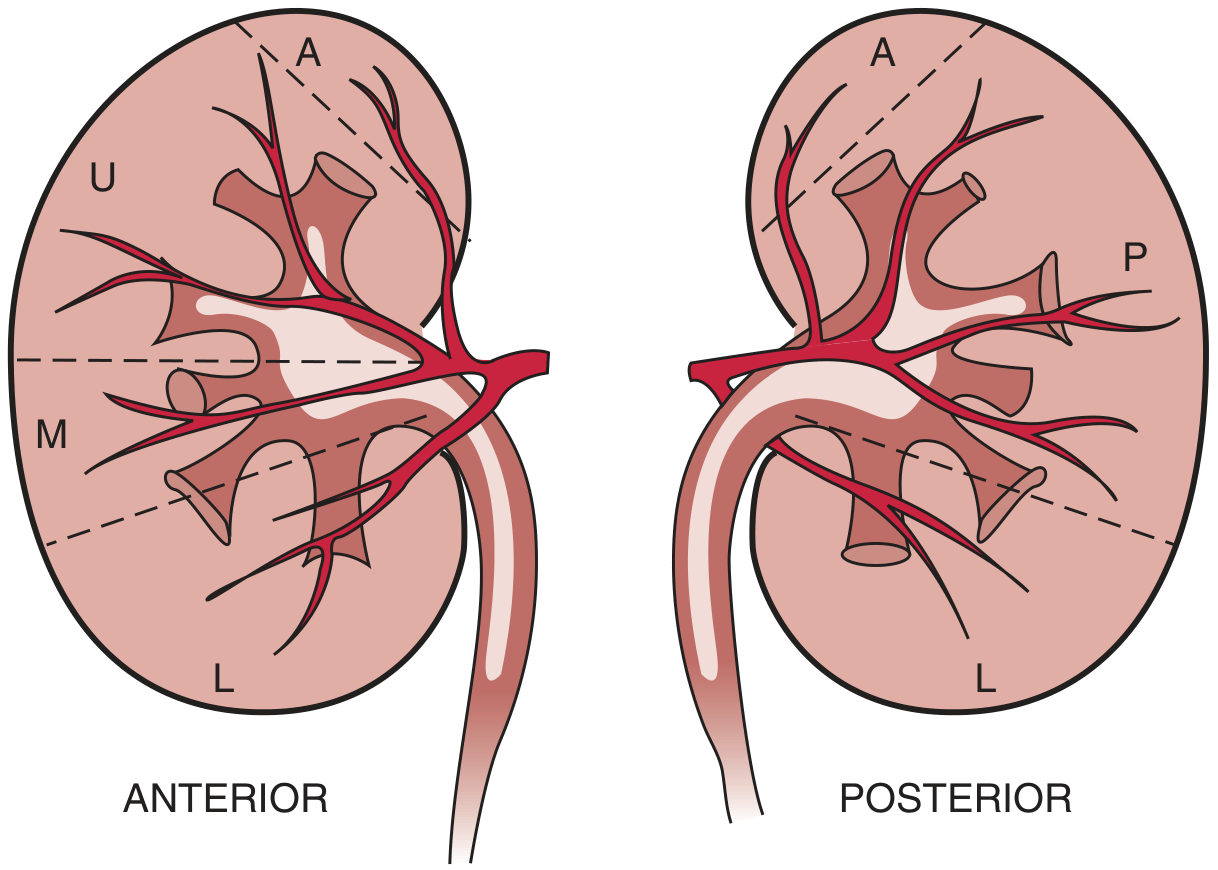
Arterial Supply - Branching Pattern
The renal artery (from the aorta at L1-L2) divides at or just before the hilum into:
Renal Artery
│
├── POSTERIOR DIVISION (first branch, arises posteriorly)
│ └── Posterior segmental artery → Posterior segment (P)
│
└── ANTERIOR DIVISION
├── Apical segmental artery → Apical segment (A)
├── Upper segmental artery → Upper segment (U)
├── Middle segmental artery → Middle segment (M)
└── Lower segmental artery → Lower segment (L)
- The posterior segmental artery is typically the first and most posterior branch, curving behind the renal pelvis
- The anterior division gives 4 branches (apical, upper, middle, lower) - variable in origin
- Accessory renal arteries (present in 25-40% of kidneys) arise directly from the aorta and may supply any segment, most commonly the lower pole
- Within each segment, the segmental arteries branch into lobar arteries → interlobar arteries (between pyramids) → arcuate arteries (at corticomedullary junction) → interlobular (cortical radiate) arteries → afferent arterioles → glomerular capillaries
Key Anatomical Principles
1. End-Arteries (No Collaterals)
Each segmental artery is a true end-artery - there are no anastomoses between segments. This has critical surgical implications:
- Clamping or ligating a segmental artery causes ischaemia and infarction of that segment only
- Allows partial (segmental) nephrectomy - removing one or more segments while preserving others
- A segment can be rendered bloodless by clamping its segmental artery before excision
2. The Avascular Intersegmental Plane (Brodel's Line / Brodel's White Line)
- Between the anterior and posterior vascular territories lies a relatively avascular longitudinal plane on the posterolateral surface of the kidney
- Known as Brodel's line (or the avascular plane of Brodel)
- Located approximately 1-2 cm posterior to the lateral convex border
- This is the optimal incision line for nephrotomy (e.g., anatrophic nephrolithotomy) - incising here minimizes bleeding
- Identification intraoperatively: The posterior segmental artery is temporarily clamped and methylene blue is given IV - the posterior segment blanches while the anterior segments turn blue, clearly delineating the avascular plane
"Clamping the posterior segmental artery will result in blanching of the posterior segment of the kidney, the blue-colored parenchyma allowing delineation of the avascular intersegmental plane, that is, the Brodel line." - Hinman's Atlas of Urologic Surgery
3. Venous Drainage Differs From Arterial Supply
- Renal veins are NOT end-vessels - they form an extensive anastomotic network
- Multiple intersegmental venous communications exist throughout the kidney
- Therefore, a renal vein can be ligated without causing segmental infarction (unlike arteries)
- This is exploited in partial nephrectomy - venous bleeding is more easily controlled
Microanatomy: Further Vascular Hierarchy Within Each Segment
| Vessel | Location | Function |
|---|---|---|
| Segmental artery | Renal sinus | Supplies one of 5 segments |
| Lobar artery | Renal sinus | One per renal lobe (pyramid + cortex) |
| Interlobar artery | Between pyramids (renal columns of Bertin) | Ascends toward cortex |
| Arcuate artery | Corticomedullary junction | Arcs parallel to surface |
| Interlobular (cortical radiate) artery | Cortex, between lobules | Perpendicular to surface |
| Afferent arteriole | Cortex | Supplies individual glomerulus |
| Glomerular capillaries | Bowman's capsule | Filtration |
| Efferent arteriole | Post-glomerular | Peritubular capillaries (cortex) or vasa recta (medulla) |
| Vasa recta | Medulla | Countercurrent exchange; concentrating mechanism |
Surgical Significance
| Application | Relevance of Segmental Anatomy |
|---|---|
| Partial nephrectomy (renal cell carcinoma, trauma) | Resect along intersegmental planes; preserve segments with intact blood supply |
| Anatrophic nephrolithotomy | Nephrotomy along Brodel's avascular line minimizes haemorrhage |
| Renal transplant | Multiple renal arteries (accessory) require separate anastomosis to avoid segmental ischaemia |
| Percutaneous nephrolithotomy (PCNL) | Posterior calyceal puncture preferred - access through relatively avascular posterior parenchyma |
| Renal trauma management | Grade V injuries - segmental vascular injuries guide angioembolization or partial resection |
| Renal angiography | Selective embolization can target individual segments (e.g., for AVM, pseudoaneurysm, haemorrhage) |
Sources:
- Brenner & Rector's The Kidney, 2-Volume Set - Chapter 2, Gross Features (Fig. 2.1; modified from Graves FT, Br J Surg 1954;42:132-139)
- Sabiston Textbook of Surgery, 21st Ed. - Chapter 29, Kidney (p. 3746)
- Hinman's Atlas of Urologic Surgery - Chapter 24, Anatrophic Nephrolithotomy (pp. 217-218)
How would you manage a 30-year-old young lady with painless enlargement of lower pole of left kidney?
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
I have comprehensive, authoritative content. Now let me compile a thorough structured clinical management answer.
Management of a 30-Year-Old Woman with Painless Enlargement of the Lower Pole of the Left Kidney
Step 1: Clinical Assessment
History
A focused history must address:
Regarding the mass:
- Duration of swelling - slow vs. rapid growth
- Any change in size
- Associated symptoms: haematuria (gross or micro), flank/back pain, fever
- History of trauma (perinephric haematoma, urinoma)
- Prior urinary tract infections or stone disease
Systemic symptoms:
- Weight loss, anorexia, fever (paraneoplastic / malignancy)
- Hypertension (renal artery compression, renin-secreting tumour)
- Polycythaemia symptoms (erythropoietin-secreting RCC)
- Hypercalcaemia symptoms (PTHrP-secreting RCC)
Relevant background:
- Family history of renal tumours (hereditary RCC syndromes: VHL, Birt-Hogg-Dubé, hereditary papillary RCC)
- History of tuberous sclerosis (angiomyolipoma)
- Smoking history (RCC risk factor)
- Obstetric history (last menstrual period - rule out pelvic mass or pregnancy-related)
- Oral contraceptive use (rare renal haemangioma/lipoma associations)
Physical Examination
- General: Pallor, cachexia, lymphadenopathy
- Abdominal examination:
- Inspect: visible flank fullness
- Palpate: bimanual ballotment of the kidney - confirm the mass is renal (ballotable, moves with respiration, cannot get above it), distinguish from splenomegaly or colonic mass (left-sided)
- Percuss: resonant anteriorly (renal mass - retroperitoneal) vs. dull (colonic)
- Left renal lower-pole mass: confirm it does not cross midline; assess mobility and consistency
- Varicocoele (left-sided): A new left-sided varicocoele that does not decompress on lying flat suggests left renal vein obstruction (e.g., by a large tumour or tumour thrombus)
- BP measurement (hypertension)
- Skin: Angiofibromas, periungual fibromas (tuberous sclerosis → angiomyolipoma)
Step 2: Differential Diagnosis
Given her age (30 years), sex, and painless enlargement, the differential is:
| Category | Diagnosis | Key Features |
|---|---|---|
| Renal cysts | Simple cyst | Commonest; smooth, non-tender; fluid on US |
| Complex cyst (Bosniak IIF-IV) | Septations, calcification, enhancement | |
| Hydronephrosis (lower-pole dilatation) | Pelviureteric junction / infundibular obstruction | |
| Benign solid tumours | Angiomyolipoma (AML) | Most common benign solid renal mass in young women; fat on CT; associated with tuberous sclerosis |
| Oncocytoma | Benign; cannot reliably distinguish from RCC on imaging; central scar on CT (not specific) | |
| Renal lipoma | Rare | |
| Multilocular cystic nephroma | Young women; second peak in 4th-5th decade | |
| Malignant | Renal cell carcinoma (RCC) - clear cell | Uncommon at 30 but must exclude; enhancing solid mass |
| Papillary RCC | More indolent; lower attenuation on CT | |
| Chromophobe RCC | Better prognosis | |
| Wilms' tumour (nephroblastoma) | Rare in adults; large mixed mass | |
| Urothelial carcinoma (renal pelvis) | More common in older patients; causes haematuria | |
| Collecting duct carcinoma | Aggressive; infiltrative pattern | |
| Renal medullary carcinoma | Associated with sickle cell trait - important in young patients | |
| Metastasis / lymphoma | Lymphoma: infiltrative, bilateral; metastasis: history of primary | |
| Vascular | Renal artery aneurysm | Calcified; pulsatile |
| Arteriovenous malformation | Rare | |
| Inflammatory | Xanthogranulomatous pyelonephritis | Usually unilateral; staghorn calculus |
| Perinephric abscess / renal abscess | Fever, toxicity | |
| Renal TB (cold abscess / autonephrectomy) | Sterile pyuria | |
| Functional | Renal pseudomass (column of Bertin, dromedary hump) | Variant; no pathology |
"Current guidelines recommend multiphase, cross-sectional imaging to ensure a systematic approach and diligent evaluation of suspected renal masses, given the large differential diagnosis and considerable overlap between benign and malignant renal lesions." - Campbell-Walsh-Wein Urology
Step 3: Investigations
Baseline Laboratory Tests
| Investigation | Purpose |
|---|---|
| FBC / CBC | Anaemia (chronic disease, haematuria), polycythaemia (erythropoietin-secreting RCC) |
| Serum urea, creatinine, eGFR | Baseline renal function (critical before any intervention) |
| LFTs | Hepatic metastases; Stauffer syndrome (non-metastatic hepatic dysfunction in RCC) |
| Serum calcium | Hypercalcaemia (paraneoplastic) |
| Uric acid, LDH | Tumour burden markers |
| Coagulation screen | Pre-operative |
| Serum electrolytes | |
| Urine dipstick + microscopy | Haematuria (gross or micro), proteinuria |
| Urine cytology | If urothelial malignancy suspected (haematuria, pelvic lesion) |
| Urine culture + AFB culture | If infection / TB suspected |
| Pregnancy test (urine/serum βhCG) | Must rule out in any 30-year-old woman with abdominal mass |
Imaging
Step A: Ultrasound (US) - First Line
- Readily available, no radiation, no contrast
- Characterizes mass as:
- Cystic (anechoic, posterior enhancement, thin wall) → Bosniak I = simple cyst
- Solid (echogenic, vascular on Doppler)
- Complex (mixed, septated)
- Detects fat within mass - strongly suggests angiomyolipoma (echogenic on US)
- Identifies lower-pole dilatation (hydronephrosis vs. solid mass)
- Can detect ipsilateral varicocoele (left side)
- Limitation: Cannot definitively characterize complex lesions or detect small masses
Step B: CT with Renal Mass Protocol (Definitive Imaging)
- Gold standard for characterizing renal masses
- Protocol: Unenhanced + arterial/corticomedullary phase + nephrographic phase + excretory (delayed) phase
- Key CT findings:
| Finding | Diagnosis |
|---|---|
| No enhancement, fluid density (<20 HU) | Simple cyst |
| Fat density (-10 to -100 HU) within mass | Angiomyolipoma (pathognomonic) |
| Strong enhancement (>20 HU increase) | Clear cell RCC, oncocytoma |
| Moderate enhancement, low attenuation | Papillary RCC |
| Central scar (not specific) | Oncocytoma (also seen in RCC) |
| Calcification, thick irregular septa | Complex cyst / cystic RCC |
| Infiltrative, bilateral | Lymphoma |
| Tumour thrombus in renal vein/IVC | RCC (vascular invasion) |
| Enlarged lymph nodes, hepatic lesions | Metastatic disease |
- CT chest, abdomen, pelvis: staging if malignancy suspected
Bosniak Classification (for cystic lesions):
| Category | Description | Malignancy Risk | Action |
|---|---|---|---|
| I | Simple thin-walled cyst, no enhancement | 0% | No follow-up needed |
| II | Hairline septa, fine calcification, no enhancement | <5% | No follow-up needed |
| IIF | Multiple thin septa, minimal wall thickening, no measurable enhancement, high-density cysts >3 cm | ~20% | 6-monthly CT/MRI for 1 year, then annual for 5 years |
| III | Thickened, irregular or smooth enhancing walls/septa | ~50% | Surgical excision or ablation |
| IV | Solid enhancing components independent of wall | ~90% | Surgical excision |
Step C: MRI
- Used when CT is indeterminate, patient has renal impairment, or iodinated contrast contraindicated
- Best for characterizing tumour thrombus extent (IVC involvement)
- Can use gadolinium to assess enhancement
- No radiation (relevant in young woman)
- Diffusion-weighted imaging (DWI) can help distinguish cyst from solid
Step D: Nuclear Medicine
- DMSA or MAG3 scan: Assesses split renal function - critical if partial or total nephrectomy is planned
- PET-CT: Not routine for primary diagnosis but useful for staging/restaging
Step 4: Renal Biopsy
Indications
- Mass suspicious for lymphoma or metastasis (where surgery is not primary treatment)
- Indeterminate imaging findings where histology will change management
- Small renal mass (<4 cm) in a patient who would be offered ablative therapy or active surveillance instead of surgery
- Patient unwilling for surgery - histology needed to counsel
- Suspected renal abscess / TB (culture from biopsy)
Considerations
- Not routinely required before nephrectomy for a clearly malignant-appearing solid enhancing mass - risk of seeding is very low but accepted practice is to operate
- Core biopsy preferred over FNA for solid masses - better histological yield
- CT-guided percutaneous approach
Step 5: Treatment - Based on Diagnosis
A. If Simple Cyst (Bosniak I-II)
- No treatment required
- Reassurance
- No follow-up imaging needed for Bosniak I/II
B. If Angiomyolipoma (AML)
- Observation if <4 cm and asymptomatic - annual US follow-up
- Embolization or nephron-sparing surgery if:
- Size ≥4 cm (risk of spontaneous haemorrhage - Wunderlich syndrome increases)
- Symptomatic (pain, bleeding)
- Rapid growth
- Important: AML in young women with tuberous sclerosis requires multidisciplinary management
C. If Renal Cell Carcinoma (or Strongly Suspected Malignant Solid Mass)
Staging (TNM)
- CT chest/abdomen/pelvis
- Bone scan if alkaline phosphatase raised or bone pain
- Brain MRI if neurological symptoms
- Isotope renal scan for split function
Surgical Treatment (Localized Disease - T1-T3)
| Tumour | Preferred Surgery |
|---|---|
| T1a (<4 cm) | Partial nephrectomy (nephron-sparing) - laparoscopic or robotic; preserves maximum renal function; strongly preferred in a 30-year-old |
| T1b (4-7 cm) | Partial nephrectomy if technically feasible; radical nephrectomy if not |
| T2 (>7 cm, organ-confined) | Radical nephrectomy (laparoscopic or open) |
| T3 (local extension / vascular involvement) | Radical nephrectomy ± IVC thrombectomy |
| T4 / M1 | Systemic therapy (TKI, immunotherapy) ± cytoreductive nephrectomy in selected cases |
In a 30-year-old woman, nephron-sparing (partial) nephrectomy is the priority - preservation of long-term renal function is paramount, and she has decades ahead of her. Radical nephrectomy is only acceptable if partial is technically not achievable.
Thermal Ablation (Radiofrequency / Cryoablation)
- Option for small tumours (<3 cm) in patients who decline surgery or have high operative risk
- Less appropriate in a fit 30-year-old as primary treatment
Active Surveillance
- Selected small (<3 cm) indolent masses in poor operative candidates
- Not typically appropriate for a fit 30-year-old
Systemic Therapy (Metastatic Disease)
- First line: Immunotherapy combinations (nivolumab + ipilimumab) or TKI + immunotherapy (pembrolizumab + axitinib, etc.)
- Targeted therapy: sunitinib, pazopanib (VEGFR inhibitors); everolimus, temsirolimus (mTOR inhibitors)
D. If Hydronephrosis (Lower-Pole Dilatation)
- Identify cause: UPJ obstruction, lower-pole infundibular stenosis, crossing vessel
- Pyeloplasty (Anderson-Hynes dismembered pyeloplasty) for UPJ obstruction - laparoscopic preferred
- Endoscopic balloon dilatation for infundibular stenosis
E. If Wilms' Tumour (Adult)
- Rare but must be considered
- Treated with radical nephrectomy + adjuvant chemotherapy (actinomycin-D, vincristine ± doxorubicin)
- Staging guides adjuvant treatment
F. If Renal Abscess / Infection
- Antibiotics + percutaneous drainage if >3 cm
- TB: standard 6-month antitubercular regimen
Summary Algorithm
30-year-old woman, painless lower-pole left renal mass
│
┌─────── HISTORY + EXAMINATION ────────┐
│ (BP, varicocoele, skin signs, │
│ family history, haematuria) │
│
PREGNANCY TEST
│
URINE dipstick + microscopy
│
BLOODS (FBC, U&E, LFTs, Ca²⁺, LDH)
│
ULTRASOUND ABDOMEN/PELVIS
│
┌──────────┼──────────────┐
│ │ │
Cystic Solid Complex/
(Bosniak I) mass indeterminate
│ │ │
Reassure CT RENAL CT RENAL MASS
No F/U MASS PROTOCOL PROTOCOL ± MRI
│ │
┌─────────┼─────────┐ │
│ │ │ │
Fat Enhancing Bosniak Bosniak
present solid mass I/II IIF/III/IV
│ │ │ │
AML RCC/ No F/U F/U or
(>4cm: Oncocytoma Surgery
embolize) │
┌─────┴─────┐
│ │
Staging Biopsy
(CT C/A/P) (if lymphoma/
│ met suspected)
│
PARTIAL NEPHRECTOMY
(preferred in young patient)
or Radical nephrectomy
Key Points Specific to a 30-Year-Old Woman
- Always exclude pregnancy before any imaging (urinary βhCG first)
- MRI is preferred over CT where possible in young women (no radiation, no renal contrast concerns) - especially if the mass is <4 cm and partial nephrectomy is planned
- Angiomyolipoma is proportionally more common in young women than RCC - fat on imaging is diagnostic
- Renal medullary carcinoma should be considered if she has sickle cell trait (aggressive; arises in renal medulla of young Black patients)
- Nephron-sparing surgery is the absolute priority - a 30-year-old has 50+ years of renal function ahead; loss of a kidney puts her at higher risk of CKD with ageing
- Hereditary syndromes should be screened for if RCC confirmed (VHL disease, Birt-Hogg-Dubé syndrome) given young age at presentation
Sources:
- Campbell-Walsh-Wein Urology - Chapter 97, Radiographic Evaluation of Renal Masses (Table 97.2, pp. 2842-2843)
- Grainger & Allison's Diagnostic Radiology - Chapter 28, Bosniak Renal Cyst Classification (Table 28.1, pp. 776-779)
- Brenner & Rector's The Kidney - Chapter 25, Renal Mass: Cysts to Renal Cell Carcinoma
Polyposis syndrome of the colon
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
I now have comprehensive, authoritative content from multiple textbooks. Let me compile the full answer.
Polyposis Syndromes of the Colon
Colonic polyposis syndromes are a heterogeneous group of conditions - hereditary or acquired - characterized by multiple colorectal polyps. They are classified primarily by polyp histology:
| Class | Syndromes |
|---|---|
| Adenomatous polyposis | FAP, AFAP, Gardner's, Turcot's, MUTYH-AP, Polymerase proofreading-AP, Lynch-associated |
| Hamartomatous polyposis | Peutz-Jeghers, Juvenile polyposis, Cowden/PTEN hamartoma, Bannayan-Riley-Ruvalcaba |
| Serrated polyposis | Serrated polyposis syndrome |
| Rare / miscellaneous | Cronkhite-Canada, Cowden, neurofibromatosis, inflammatory polyposis |
I. Adenomatous Polyposis Syndromes
1. Familial Adenomatous Polyposis (FAP)
Genetics
- Autosomal dominant, near-complete penetrance (each child of an affected parent has 50% risk)
- Germline mutation in the APC (adenomatous polyposis coli) tumour suppressor gene, chromosome 5q21
- APC is a two-hit tumour suppressor: the inherited mutant allele + a somatic second-hit event in individual cells → adenoma initiation
- Up to 20% are de novo mutations without family history
- ~25% of patients test negative for APC mutation → often represents AFAP or MAP
- Prevalence: 1 in 6,850 to 1 in 31,250 live births
Colonic Manifestations
- Hundreds to thousands (100-10,000+) of adenomatous polyps throughout the entire colorectum
- Polyps typically appear in late teens to early 20s
- Colorectal cancer (CRC) is inevitable without prophylactic colectomy
- Average age at CRC diagnosis: 39 years (St Mark's series)
- By age 45: 87% have CRC; by age 50: 93%
- ~48% have multiple synchronous CRCs at diagnosis
- Cancer onset as early as age 9 reported (though rare before adolescence)
Extracolonic Manifestations (Classic FAP)
Upper GI:
- Gastric fundic gland polyps (23-100% of patients) - hamartomas; usually benign but may dysplase
- Duodenal adenomas (>50%) - particularly periampullary; 2nd highest cancer risk in FAP
- Periampullary adenocarcinoma in 3-10% - leading cause of cancer death after colectomy
- Gastric cancer lifetime risk: ~0.6%
Soft tissue / skeletal:
- Desmoid tumours (~10%) - fibromatous lesions of myofibroblasts in mesentery, abdominal wall, extremities; locally invasive (low-grade sarcoma-like); lethal in ~10%; 3rd most common cause of FAP mortality
- Epidermoid (sebaceous) cysts
- Lipomas, fibromas
Skeletal:
- Osteomas (mandible, skull, long bones) - Gardner's syndrome feature
- Dental anomalies (supernumerary/impacted teeth)
Ophthalmic:
- Congenital hypertrophy of the retinal pigment epithelium (CHRPE) - bilateral, multiple; an early, non-progressive marker; detected before polyps emerge
CNS:
- Brain tumours (medulloblastoma) - Turcot's syndrome overlap
2. Gardner Syndrome
- Variant of FAP (same APC gene mutation) - now considered part of the FAP spectrum
- Defined by prominent extracolonic manifestations:
- Osteomas (mandible, skull)
- Epidermoid/sebaceous cysts
- Desmoid tumours
- Thyroid neoplasms
- CHRPE
- Colonic polyp phenotype identical to classic FAP
- Expression of extracolonic features correlates with location of APC mutation (mutations at codons 1310-2011 associated with desmoids; codons 1250-1330 with more severe polyposis)
- Term is mainly of historical interest but still used clinically when extraintestinal manifestations are prominent
3. Turcot Syndrome
- Characterized by colonic polyposis + CNS tumours
- Two genetically distinct entities:
- Type 1 (~2/3): APC mutation → classic colonic polyposis + medulloblastoma (cerebellar); risk 92× general population
- Type 2 (~1/3): Mismatch repair (MMR) gene mutation (Lynch syndrome) → fewer adenomas + glioblastoma multiforme
- Also: anaplastic astrocytomas, ependymomas
4. Attenuated FAP (AFAP)
- Milder FAP variant from APC mutations at far 5' or 3' ends, or exon 9
- Features:
- Fewer polyps: average 25 (range 0-470), typically 10-100
- Right-sided colonic predominance (vs. pan-colonic in FAP)
- Later onset: CRC typically at ~58 years (range 29-81)
- CRC risk up to 69-80% by age 80 - still significant
- Extracolonic features: similar to FAP; upper GI polyps NOT attenuated
- Some patients manageable endoscopically (avoid colectomy if colon can be cleared); most need colectomy with IRA
- Difficult to diagnose without genetic testing (can present with very few polyps)
5. MUTYH-Associated Polyposis (MAP)
- Only autosomal recessive polyposis syndrome
- Biallelic germline mutations of MUTYH (MYH) - a base excision repair gene
- Loss of MUTYH causes G:C → T:A transversions → somatic APC and KRAS mutations
- Prevalence: accounts for <1% of all CRC; found in 7.5-12.5% of patients with >100 adenomas who are APC-negative
- Phenotype: attenuated polyposis (10-100s of adenomas); mixed adenomatous and serrated polyp histology
- CRC risk: up to 80% lifetime
- Management: colonoscopic surveillance; colectomy when polyps cannot be controlled endoscopically
- Genetic testing: must test both alleles (recessive); siblings have 25% risk
6. Polymerase Proofreading-Associated Polyposis (PPAP)
- Mutations in POLE and POLD1 genes (DNA polymerase proofreading)
- Phenotype overlaps with AFAP/MAP
- Autosomal dominant; associated with early-onset CRC and endometrial cancer
II. Hamartomatous Polyposis Syndromes
7. Peutz-Jeghers Syndrome (PJS)
Genetics
- Autosomal dominant; germline mutation in STK11 (LKB1), chromosome 19p13.3
- STK11 is a serine/threonine kinase tumour suppressor gene
Features
Pathognomonic triad:
- Mucocutaneous melanin pigmentation - dark brown/black macules on lips, buccal mucosa, perioral skin, nostrils, perianal region; present in childhood but may fade in adulthood
- Hamartomatous polyps throughout the GI tract (stomach, small bowel > large bowel); polyps have smooth muscle core (arborizing pattern) distinguishing them from juvenile polyps
- Elevated cancer risk
GI complications:
- Small bowel intussusception (classic) - episodic colicky pain, potentially life-threatening
- Bleeding and anaemia from polyps
- Obstruction
Cancer risks (lifetime):
| Site | Risk |
|---|---|
| Colorectal | 39% |
| Stomach | 29% |
| Small bowel | 13% |
| Pancreas | 11-36% (very high) |
| Breast | 45-50% (major risk) |
| Ovary | 21% |
| Cervix (adenoma malignum) | 10% |
| Sertoli cell testicular tumour (males) | 9% |
Management: Surveillance endoscopy (upper + lower + capsule endoscopy for small bowel); breast screening; pancreatic surveillance; gynaecological surveillance; prophylactic mastectomy/salpingo-oophorectomy considered after childbearing
8. Juvenile Polyposis Syndrome (JPS)
Genetics
- Autosomal dominant
- Mutations in SMAD4 (18q21) or BMPR1A (10q22) - TGF-β/BMP signalling pathway
- Each accounts for ~20% of JPS; ~60% have no identifiable mutation
- ~25% de novo mutation rate
Key Points
- "Juvenile" refers to polyp histology (cystic glands, oedematous lamina propria, inflammatory infiltrate), NOT patient age
- Diagnosis: >5 juvenile polyps in colon, OR multiple polyps throughout GI tract, OR any juvenile polyps with positive family history
- Polyps in stomach, small intestine, and colon; range greatly in number
- CRC risk significantly elevated (lifetime risk ~40-68%)
- SMAD4 mutation is associated with JPS + hereditary haemorrhagic telangiectasia (HHT) - AVMs in lungs, liver, GI tract; presenting as epistaxis
Management: Regular colonoscopy with polypectomy; upper endoscopy; colectomy if polyps uncontrollable or CRC develops
9. Cowden Syndrome / PTEN Hamartoma Tumour Syndrome (PHTS)
Genetics
- Autosomal dominant
- PTEN (phosphatase and tensin homologue) tumour suppressor gene, chromosome 10q23
- Also: KILLIN, SDHB, SDHD mutations
Features
- Multiple benign and malignant tumours
- GI: hamartomatous and adenomatous polyps, lipomas, ganglioneuromas
- Major cancer risks:
- Breast cancer (lifetime risk 25-50%) - most clinically significant
- Thyroid cancer (follicular) - 3-10%
- Endometrial cancer - 5-10%
- Renal cancer; melanoma
- Characteristic benign lesions: trichilemmomas (facial), mucocutaneous papillomas, macrocephaly, thyroid nodules, uterine fibroids, lipomas
III. Serrated Polyposis Syndrome (SPS)
- Previously called hyperplastic polyposis syndrome
- Diagnostic criteria (WHO 2010): ≥5 serrated polyps proximal to sigmoid colon, 2+ of which ≥10 mm; OR any serrated polyps proximal to sigmoid colon with ≥1 first-degree relative with SPS; OR >20 serrated polyps of any size throughout the colon
- Polyp types: sessile serrated adenomas/lesions (SSA/SSL), hyperplastic polyps, traditional serrated adenomas
- Molecular: BRAF mutations, MLH1 promoter methylation, microsatellite instability - the "serrated pathway" to CRC
- CRC risk: significantly elevated (~7%)
- Management: annual colonoscopy with polypectomy; colectomy if uncontrollable
IV. Non-Hereditary / Acquired Polyposis Syndromes
10. Cronkhite-Canada Syndrome (CCS)
- Non-hereditary, acquired (no germline mutation)
- Rare, typically older adults; aetiology unknown (possibly autoimmune)
- Features:
- GI hamartomatous polyposis (stomach to rectum)
- Ectodermal triad: alopecia + onychodystrophy (nail changes) + hyperpigmentation
- Protein-losing enteropathy, diarrhoea, weight loss
- No family history - distinguishes from inherited syndromes
- Poor prognosis; mortality ~55%; responds to steroids, nutritional support, antibiotics
V. Surgical Management of FAP (Summary)
Four operations are available; choice depends on extent of rectal polyposis, presence of cancer, desmoid risk, and patient preference:
| Operation | Indication | Pros | Cons |
|---|---|---|---|
| Total proctocolectomy + end ileostomy (Brooke) | Rectal cancer, poor sphincter, desmoid preventing IPAA | Complete cancer removal | Permanent stoma |
| Restorative proctocolectomy + IPAA | Standard preferred operation; diffuse polyposis | No stoma; preserves continence | Pouch complications; residual anal transition zone at risk; surveillance needed |
| Total abdominal colectomy + ileorectal anastomosis (IRA) | <20-30 rectal polyps manageable endoscopically; classic FAP | Simpler; better function | Residual rectum at cancer risk; 6-12 monthly surveillance mandatory |
| Continent ileostomy (Kock pouch) | Patients who want no external appliance | No external appliance | Complex; high revision rate |
Key surgical timing: Ideally after physical maturity; polyp burden and symptoms guide urgency.
Medical adjuncts:
- COX-2 inhibitors (celecoxib, sulindac) - reduce polyp burden; adjunct, NOT substitute for surgery
- Desmoid management: NSAIDs, tamoxifen (hormone-responsive), imatinib; surgery avoided due to local recurrence
Surveillance after surgery:
- IRA: flexible endoscopy every 6-12 months
- Ileal pouch: pouchoscopy every 1-3 years
- Upper GI endoscopy: every 1-3 years from age 25-30 (duodenal/periampullary surveillance)
Summary Comparison Table
| Syndrome | Gene | Inheritance | Polyp Type | CRC Risk | Distinguishing Feature |
|---|---|---|---|---|---|
| FAP | APC | AD | Adenoma (100-1000s) | ~100% (by 45) | Diffuse adenomatosis; inevitable CRC |
| AFAP | APC | AD | Adenoma (10-100) | ~69-80% | Right-sided; later onset |
| Gardner's | APC | AD | Adenoma | ~100% | + Osteomas, desmoids, cysts |
| Turcot's | APC / MMR | AD | Adenoma | ~100% | + Medulloblastoma (APC) / GBM (MMR) |
| MAP | MUTYH | AR | Mixed adenoma/serrated | ~80% | Only AR polyposis; APC-negative FAP-like |
| Peutz-Jeghers | STK11 | AD | Hamartoma | 39% CRC; 50% breast | Mucocutaneous pigmentation; intussusception |
| Juvenile polyposis | SMAD4/BMPR1A | AD | Hamartoma (juvenile) | 40-68% | + HHT if SMAD4; cystic stroma |
| Cowden/PHTS | PTEN | AD | Hamartoma | Modest CRC; 50% breast | Trichilemmomas; thyroid; breast |
| Serrated polyposis | Unknown | Variable | Serrated | ~7% | Proximal, large serrated polyps |
| Cronkhite-Canada | None (acquired) | Non-hereditary | Hamartoma | Moderate | Alopecia, nails, pigmentation |
Sources:
- Yamada's Textbook of Gastroenterology, 7th Ed. - Chapter 71, Polyposis Syndromes; Chapter on Hamartomatous Polyposis
- Maingot's Abdominal Operations - Chapter: Familial Adenomatous Polyposis
- Schwartz's Principles of Surgery, 11th Ed. - Inherited Colorectal Carcinoma
- Mulholland & Greenfield's Surgery, 7th Ed. - Surgical Treatment of FAP (Table 67.1)
This is a shared conversation. Sign in to Orris to start your own chat.