Cystic fibrosis
Reading File
Asking for Preferences
Finding Sources
Searching PubMed
"cystic fibrosis" AND diagnosis
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive, well-sourced information. Here is a thorough overview of cystic fibrosis diagnosis and investigations.
Cystic Fibrosis: Diagnosis & Investigations
Diagnostic Criteria (CFF Consensus Guidelines)
A diagnosis of CF requires both of the following:
Criterion 1 — One or more of:
- Characteristic phenotypic features (sinopulmonary disease, GI/nutritional abnormalities, salt loss, male urogenital abnormalities)
- Family history of CF in a first-degree relative
- Positive newborn screening (NBS) result
Criterion 2 — Evidence of CFTR dysfunction via at least one of:
- Sweat chloride ≥60 mmol/L (diagnostic)
- Sweat chloride in the intermediate range (30–59 mmol/L) plus identification of two disease-causing CFTR mutations
- Sweat chloride in the intermediate range plus demonstration of abnormal nasal epithelial ion transport (nasal potential difference or intestinal current measurement)
(Robbins Pathologic Basis of Disease; CFF Guidelines, Farrell et al. J Pediatr 2017)
Diagnostic Algorithm
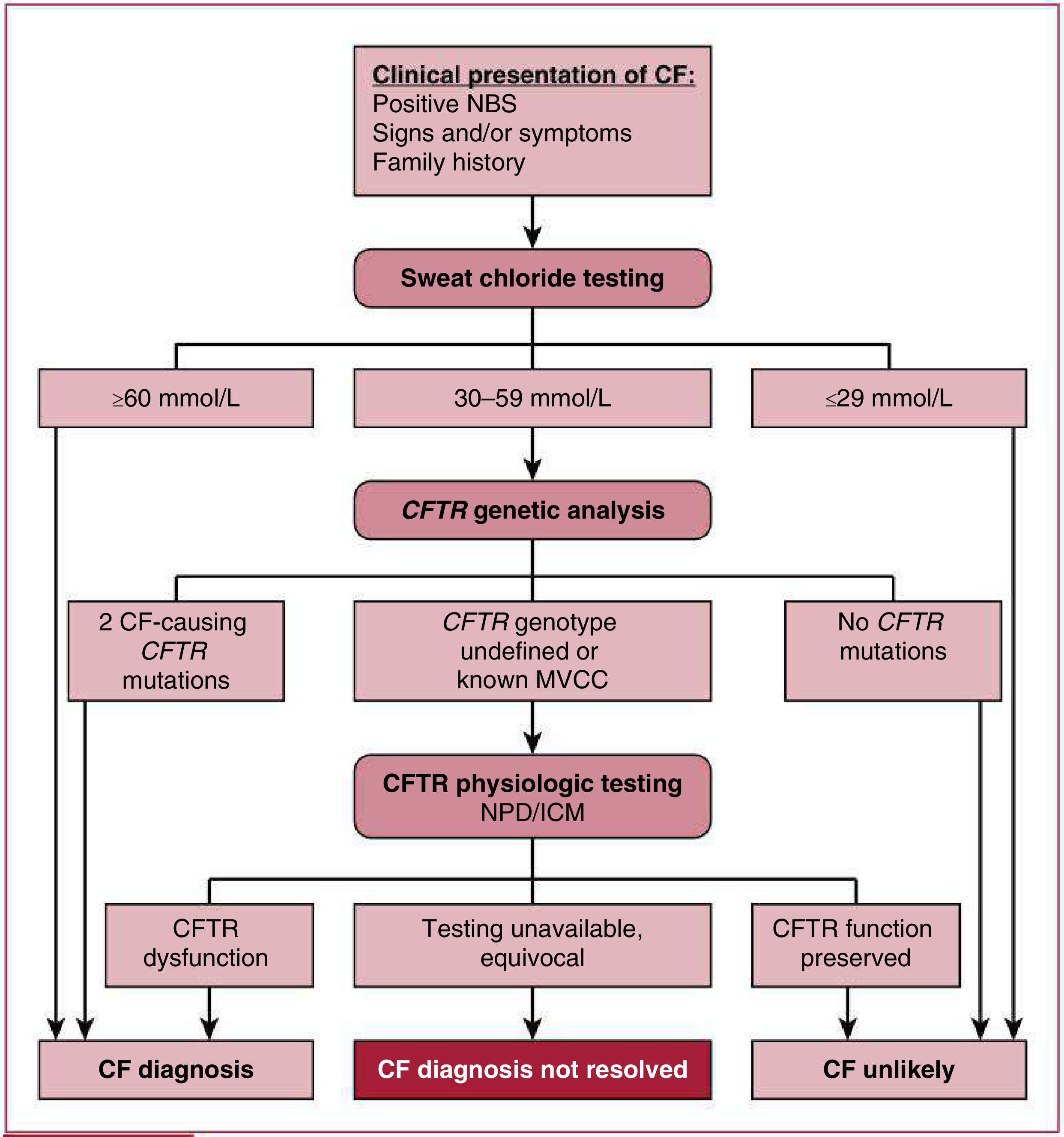
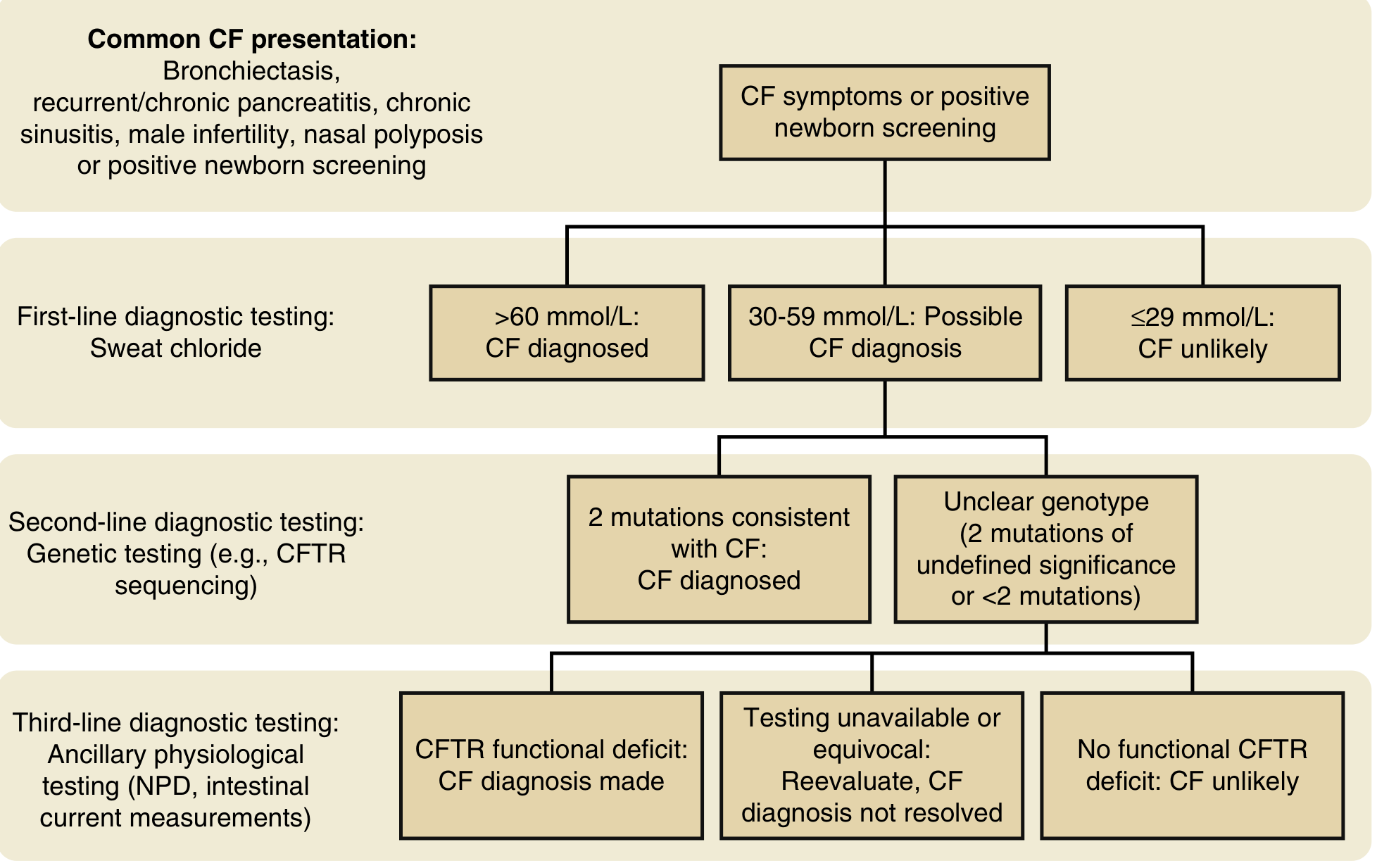
1. Newborn Screening (NBS)
- The most common NBS test measures immunoreactive trypsinogen (IRT) in blood — elevated because pancreatic injury in CF causes trypsinogen to leak into circulation.
- Over 60% of CF patients in the USA are now diagnosed via NBS (median age at diagnosis: 4 months).
- A positive NBS is presumptive only — must be confirmed with sweat chloride testing ± genetic analysis.
- Sleisenger & Fordtran's GI & Liver Disease
2. Sweat Chloride Testing — The Gold Standard
Performed by quantitative pilocarpine iontophoresis (QPIT):
| Sweat Cl⁻ (mmol/L) | Interpretation |
|---|---|
| ≥60 | CF diagnostic |
| 30–59 | Intermediate — further workup required |
| ≤29 | CF unlikely |
Key points:
- Updated CFF guidance lowered the intermediate range threshold from 40 to 30 mmol/L, improving detection of milder/atypical CF without increasing false positives.
- The test should be performed at a certified CF center using standardized methods — false positives and false negatives occur with malnutrition, certain medications, or inadequate sweat volume.
- Conditions other than CF that raise sweat electrolytes include ectodermal dysplasia, adrenal insufficiency, glycogen storage disease type 1, familial hypoparathyroidism, and mucopolysaccharidoses.
- Harrison's Principles of Internal Medicine 22E; Sleisenger & Fordtran's
Indications for sweat testing include: siblings of CF patients, chronic pulmonary symptoms, bronchiectasis, recurrent respiratory infections, failure to thrive, nasal polyposis, meconium ileus, rectal prolapse, jaundice in early infancy, cirrhosis in childhood, male aspermia/azoospermia, and heat stroke.
3. CFTR Genetic Analysis (Second-Line)
- Triggered by an intermediate sweat chloride (30–59 mmol/L) or when confirmation is needed.
- Full CFTR sequencing (or high-sensitivity panel) is preferred — standard panels testing <40 mutations are insufficient for atypical/mild cases, as they are biased toward common Caucasian mutations and will miss rare partial-function variants.
- Two CF-causing mutations = CF confirmed.
- One or zero mutations with intermediate sweat Cl⁻ → proceed to third-line physiologic testing.
- Genotype information is catalogued in the CFTR-2 repository (cftr2.org), useful for predicting phenotypic severity and guiding mutation-specific therapy.
- Murray & Nadel's Textbook of Respiratory Medicine; Thompson & Thompson Genetics 9e
4. CFTR Physiologic Testing (Third-Line)
Used when sweat Cl⁻ is intermediate and genetic testing is unclear/insufficient:
| Test | Principle | Availability |
|---|---|---|
| Nasal potential difference (NPD) | Measures transepithelial ion transport via nasal epithelium; abnormal in CF | Many CF research centers |
| Intestinal current measurement (ICM) | Measures CFTR activity in rectal mucosal biopsy | Very limited; few centers worldwide |
These tests reflect CFTR functional deficits along a continuum and can distinguish mild from severe phenotypes even beyond genotype information alone.
5. Ancillary Investigations
| Test | Role |
|---|---|
| Fecal elastase-1 | Supports diagnosis by indicating pancreatic exocrine insufficiency; low in pancreatic-insufficient CF |
| Chest X-ray / CT chest | Hyperinflation, bronchiectasis, mucus plugging, air trapping |
| Pulmonary function tests (spirometry) | Obstructive pattern (↓FEV₁, ↓FEV₁/FVC); tracks disease progression |
| Sputum culture | Identifies organisms — S. aureus (early), P. aeruginosa (chronic), B. cepacia (poor prognosis) |
| Abdominal imaging / MRCP | Assesses hepatobiliary disease, pancreatic changes |
| Semen analysis | Obstructive azoospermia in ~95% of males with CF (CBAVD) |
| Liver function tests | Monitor for CF-related liver disease (onset typically around puberty) |
| OGTT / HbA1c | Screen for CF-related diabetes (up to 50% of adults) |
CFTR-Related Spectrum
CF diagnosis exists on a continuum:
- Classic CF (pancreatic-insufficient): most severe; biallelic severe CFTR mutations; sweat Cl⁻ ≥60
- Atypical/nonclassic CF: milder; often one severe + one mild mutation; may have normal/borderline sweat Cl⁻
- CFTR-related metabolic syndrome (CRMS) / CFSPD: positive NBS + intermediate sweat Cl⁻ + <2 CF-causing mutations; not CF but at risk
- CFTR-related disorders (CFTR-RD): isolated manifestations (CBAVD, idiopathic pancreatitis, bronchiectasis) in patients who don't meet full CF criteria but carry CFTR mutations
Important Caveats
- <2% of patients with classic CF phenotype have normal sweat chloride — molecular CFTR analysis is essential in these cases.
- Some patients exhibit CF-like disease without identifiable CFTR mutations — may be due to mutations in the epithelial sodium channel (ENaC) gene or non-coding CFTR regions.
- Rare CFTR mutations not on standard panels are enriched in patients with atypical/adult-onset CF — always use full sequencing in this population.
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Harrison's Principles of Internal Medicine 22E; Murray & Nadel's Textbook of Respiratory Medicine; Sleisenger & Fordtran's GI & Liver Disease; Thompson & Thompson Genetics & Genomics in Medicine 9e; Harriet Lane Handbook 23e
New treatment guidelines for tuberculosis meningitis
Reading File
Finding Sources
Finding Sources
Searching PubMed
"tuberculous meningitis" AND treatment
Searching the Web
tuberculous meningitis treatment guidelines 2024 2025
Reading File
Reading File
Reading Article
Reading Article
Reading Article
Reading Web Page
tuberculous meningitis treatment recommendations rifampin dexamethasone duration drug resistant
Reading File
Finding Sources
I now have comprehensive, current data from multiple authoritative sources including the landmark 2026 Lancet Infectious Diseases guideline. Here is the full synthesis:
Tuberculous Meningitis: New Treatment Guidelines
Overview
Tuberculous meningitis (TBM) is the most severe form of tuberculosis, causing death or disability in approximately half of all affected patients. Until 2026, there were no up-to-date international guidelines. The Tuberculous Meningitis International Research Consortium (with representatives from India, Indonesia, South Africa, Uganda, Vietnam, Australia, Netherlands, UK, and USA) published a landmark clinical practice guideline in Lancet Infectious Diseases (Feb 2026) — the most authoritative current guidance. [PMID: 40840485]
1. Anti-Tuberculosis Chemotherapy
Standard Drug-Susceptible TBM (Adults)
The backbone remains an intensive 4-drug induction regimen (2 months) followed by a continuation phase:
| Phase | Drugs | Duration |
|---|---|---|
| Intensive | Isoniazid (INH) + Rifampicin (RIF) + Pyrazinamide (PZA) + Ethambutol (EMB) | 2 months |
| Continuation | Isoniazid + Rifampicin | 7–10 months |
| Total | 12 months (WHO-recommended) |
Standard adult doses:
- Isoniazid: 300 mg/day (5 mg/kg) + pyridoxine 50 mg/day (to prevent peripheral neuropathy)
- Rifampicin: 10 mg/kg/day (max 600 mg)
- Pyrazinamide: weight-based (1000–2000 mg/day)
- Ethambutol: 15–25 mg/kg/day
Key points on duration:
- WHO recommends 12 months total; there is currently no evidence supporting longer durations producing better outcomes.
- The SURE trial (ISRCTN40829906) comparing a 6-month intensive regimen vs. 12-month standard for childhood TBM is expected to report results by end of 2025/early 2026.
- Relapse is uncommon regardless of regimen; most deaths occur in the first 6 months.
- Harrison's Internal Medicine 22E; 2026 Lancet Infect Dis Guideline
Note on ethambutol: CNS penetration is variable and questionably adequate. It can be discontinued once drug susceptibility is confirmed to isoniazid, rifampicin, and pyrazinamide. Some centres substitute ethionamide (better CNS penetration) or an aminoglycoside in the initial regimen.
Paediatric TBM — The Cape Town Regimen
The 2026 Lancet guideline and the WHO specifically recommend the Cape Town regimen for children (also adopted by South Africa's 2024 National Guidelines):
- Single phase, 6–9 months, once daily × 7 days/week
- Elevated doses to compensate for poor CNS penetration:
- Isoniazid: 15–20 mg/kg/day (max 450 mg)
- Rifampicin: 22.5–30 mg/kg/day (max 900 mg)
- Pyrazinamide: 35–45 mg/kg/day (max 2 g)
- Ethionamide: 17.5–22.5 mg/kg/day (max 1 g) — used instead of ethambutol for better CNS penetration
- 2024 SA NDoH Paediatric TB Guideline; 2026 Lancet Guideline
2. High-Dose Rifampin — NOT Recommended (New Evidence)
A critical question has been whether high-dose rifampin (35 mg/kg vs standard 10 mg/kg) improves outcomes given rifampin's limited CNS penetration.
NEJM Phase III RCT (2025) — PMID 41406445:
- 499 adults (61% HIV-positive) in Indonesia, South Africa, Uganda
- High-dose rifampin (35 mg/kg/day) vs standard (10 mg/kg/day) for 8 weeks
- Result: No benefit. 6-month mortality was 44.6% (high-dose) vs 40.7% (standard); HR 1.17 (95% CI 0.89–1.54; P = 0.25)
- Drug-induced liver injury was higher with high-dose rifampin (8.0% vs 4.4%)
- Conclusion: High-dose rifampin cannot be recommended for TBM
⚠️ This negates earlier promising phase II data and should update clinical practice.
3. Adjunctive Corticosteroids — Revised Recommendations
HIV-Negative Patients
Dexamethasone is recommended (established since the Thwaites 2004 NEJM trial):
- Dose: 0.4 mg/kg/day (or 12–16 mg/day) IV for 3–4 weeks, then taper over 3 weeks
- Reduces mortality and severe disability in HIV-negative adults and adolescents
HIV-Positive Patients — Landmark Change
NEJM RCT (2023) — PMID 37819954 (ACT HIV trial):
- 520 HIV-positive adults with TBM (Vietnam, Indonesia); double-blind, placebo-controlled
- Dexamethasone vs placebo for 6–8 weeks
- Result: No survival benefit. Mortality: 44.1% (dexamethasone) vs 49.0% (placebo); HR 0.85 (95% CI 0.66–1.10; P = 0.22)
- No subgroup demonstrated clear benefit; serious adverse events were similar
⚠️ This is a practice-changing finding. Dexamethasone should not be routinely used in HIV-positive patients with TBM. The 2026 Lancet guideline incorporates this finding.
The 2026 Lancet guideline adopts a GRADE-based approach to corticosteroids:
- Evidence-based recommendation: dexamethasone for HIV-negative TBM
- No clear recommendation for universal use in HIV-positive TBM
4. Drug-Resistant TBM
Multidrug-Resistant TBM (MDR-TBM)
- Mortality from MDR-TBM (resistant to rifampicin + isoniazid) exceeds 70%
- Poor outcomes driven by delayed resistance detection and uncertain CNS efficacy of second-line drugs
- No RCTs currently exist to guide MDR-TBM treatment
- The 2026 guideline provides good practice points (expert opinion only):
- Detect resistance early — use rapid molecular testing (Xpert MTB/RIF)
- Switch to second-line regimens incorporating fluoroquinolones (levofloxacin, moxifloxacin) — excellent CNS penetration
- Consider linezolid, bedaquiline, and other Group A/B MDR drugs, though CNS data are limited
- PET imaging studies with radiolabelled antibiotics are providing emerging insights on CNS drug distribution
Isoniazid-Monoresistant TBM
- High-dose rifampin + levofloxacin showed a survival benefit in a phase III study in isoniazid-resistant TBM (Heemskerk et al., 2016) — fluoroquinolones may replace INH in this scenario
- Goodman & Gilman's Pharmacological Basis of Therapeutics
5. Fluoroquinolones
- Excellent CNS penetration (unlike ethambutol and streptomycin)
- Levofloxacin/moxifloxacin: Added to initial regimens by many experts, especially when drug susceptibility is uncertain or in isoniazid-resistant cases
- Moxifloxacin is a Group A drug for MDR-TB pulmonary disease; its role in TBM is promising but data remain limited
- The AAP Red Book recommends adding a fluoroquinolone to initial empiric regimens for suspected TBM, particularly when susceptibility is unknown
6. Neurocritical & Neurosurgical Care
| Complication | Management |
|---|---|
| Raised intracranial pressure / hydrocephalus | External ventricular drain (EVD) or endoscopic third ventriculostomy (ETV); ventriculoperitoneal shunt (VPS) once CSF is sterile |
| SIADH | Fluid restriction; monitor sodium carefully |
| Cerebral salt-wasting | IV sodium replacement; differs from SIADH |
| Paradoxical reactions / IRIS | Corticosteroids; infliximab (TNF-inhibitor) for severe/refractory cases — case series support use [J Pediatric Infect Dis Soc 2021; Open Forum Infect Dis 2025] |
7. Antiretroviral Therapy in HIV-Co-infected Patients
- Start ART promptly — but timing relative to anti-TB treatment must account for immune reconstitution inflammatory syndrome (IRIS) risk, which can paradoxically worsen TBM
- Expert opinion favours ART initiation after clinical stabilisation (typically 2–8 weeks after TB treatment), though evidence is limited
- The 2026 guideline provides guidance on ART timing as part of its neurocritical care recommendations
Summary: What's New in 2026
| Area | Previous Practice | New Guidance |
|---|---|---|
| Corticosteroids in HIV+ TBM | Widely used | Not recommended (ACT HIV RCT, NEJM 2023) |
| High-dose rifampin | Investigated as promising | No benefit; potential harm (NEJM Phase III RCT, 2025) |
| Paediatric regimen | 2HRZ/E + 7HR | Cape Town regimen (higher doses, ethionamide, 6–9 months) |
| Treatment duration | Often extended to 18–24 months | 12 months standard; no evidence for longer |
| MDR-TBM | Poorly defined | Fluoroquinolones + early resistance detection; infliximab for IRIS |
| Comprehensive guideline | None (previous guidelines outdated) | 2026 Lancet Infect Dis GRADE-based international guideline |
Sources: Donovan J et al. A clinical practice guideline for tuberculous meningitis. Lancet Infect Dis 2026 [PMID 40840485]; Donovan J et al. Adjunctive dexamethasone for TBM in HIV-positive adults. NEJM 2023 [PMID 37819954]; Meya DB et al. Trial of high-dose oral rifampin in TBM. NEJM 2025 [PMID 41406445]; Harrison's Principles of Internal Medicine 22E; Rosen's Emergency Medicine; Goodman & Gilman's Pharmacological Basis of Therapeutics; 2024 SA NDoH Paediatric TB Guideline
Give details about cystic fibrosis diagnosis, treatment, management
Finding Sources
Finding Sources
Searching PubMed
"cystic fibrosis" AND ("CFTR modulator" OR elexacaftor OR management)
Searching the Web
cystic fibrosis treatment guidelines 2024 2025 CFTR modulators Trikafta
Reading File
Reading File
Finding Sources
Searching PubMed
"cystic fibrosis" AND ("elexacaftor tezacaftor ivacaftor" OR "vanzacaftor")
Reading File
Reading Article
Reading Article
Reading Article
Loading Image
Loading Image
Reading File
Cystic Fibrosis: Diagnosis, Treatment & Management
Pathophysiology — The Basis for Treatment
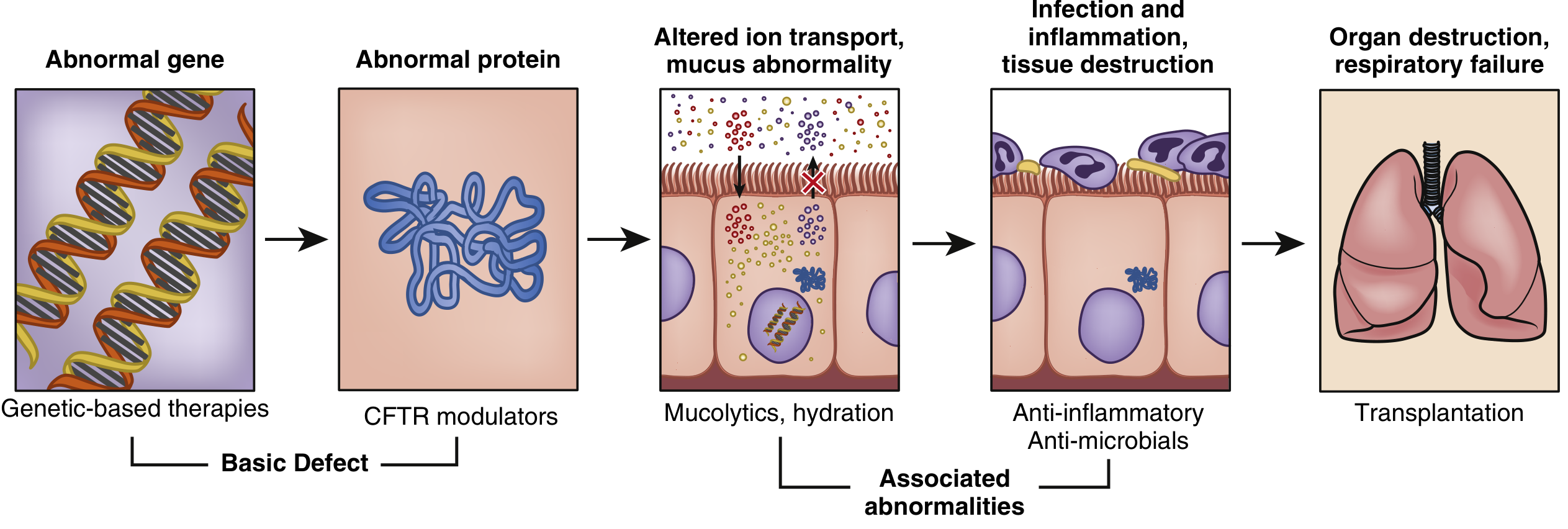
CF results from mutations in the CFTR gene (chromosome 7q31), encoding a regulated chloride channel in apical epithelial membranes. Loss of CFTR function → dehydrated, viscous mucus → impaired mucociliary clearance → chronic infection, inflammation, and progressive bronchiectasis. Treatment targets each step in this cascade.
Part 1 — Diagnosis
Diagnostic Criteria (CFF Consensus Guidelines)
Both criteria must be fulfilled:
Criterion 1 — One or more of:
- Characteristic phenotypic features (sinopulmonary disease, GI/nutritional abnormalities, salt-loss syndromes, male urogenital abnormalities)
- Family history of CF in a first-degree relative
- Positive newborn screening (NBS) test
Criterion 2 — Evidence of CFTR dysfunction:
- Sweat chloride ≥60 mmol/L, OR
- Sweat chloride 30–59 mmol/L (intermediate) + two CF-causing CFTR mutations, OR
- Sweat chloride intermediate + abnormal nasal potential difference (NPD) or intestinal current measurement (ICM)
The Diagnostic Algorithm
Tier 1 — Newborn Screening
- Immunoreactive trypsinogen (IRT) in blood — elevated due to pancreatic injury
- Over 60% of US patients now diagnosed this way (median age: 4 months)
- Positive NBS must be confirmed with sweat chloride testing
Tier 2 — Sweat Chloride Testing (Gold Standard)
| Sweat Cl⁻ | Interpretation |
|---|---|
| ≥60 mmol/L | CF diagnostic |
| 30–59 mmol/L | Intermediate — further testing required |
| ≤29 mmol/L | CF unlikely |
Performed by quantitative pilocarpine iontophoresis (QPIT) at a certified CF center.
Tier 3 — CFTR Genetic Analysis
- Triggered by intermediate sweat chloride
- Full CFTR sequencing preferred over limited panels (which miss rare mutations)
- Two CF-causing mutations = diagnosis confirmed
- Reference: CFTR-2 database for genotype-phenotype correlation
Tier 4 — CFTR Physiologic Testing
- Nasal potential difference (NPD): available at CF research centres
- Intestinal current measurement (ICM): requires rectal biopsy; very limited availability
Disease Spectrum
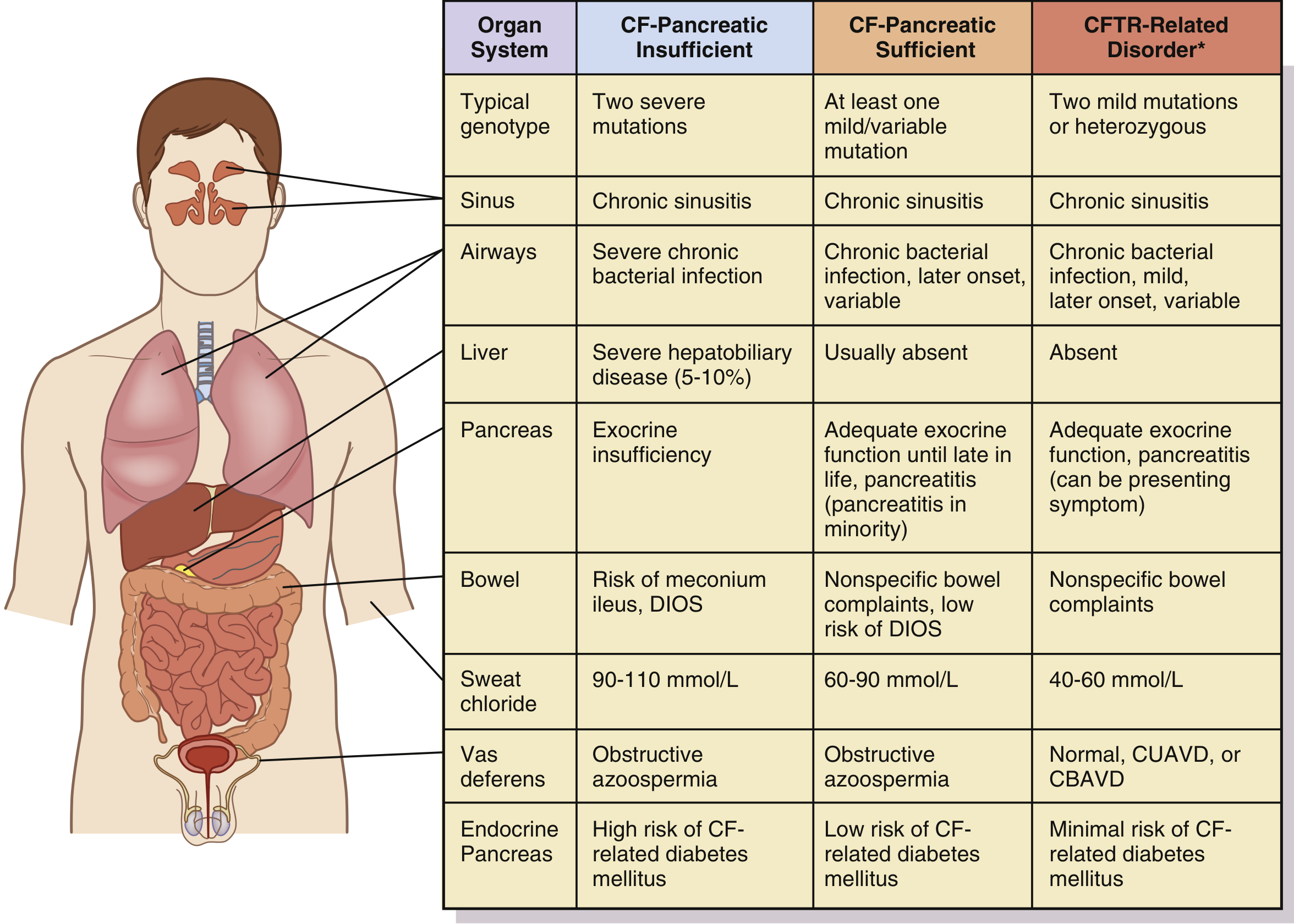
| Phenotype | Genotype | Sweat Cl⁻ | Key Features |
|---|---|---|---|
| Classic CF – pancreatic insufficient | Two severe mutations | 90–110 mmol/L | Severe pulmonary/GI disease, azoospermia |
| Classic CF – pancreatic sufficient | One mild/variable mutation | 60–90 mmol/L | Pulmonary disease, pancreatitis risk |
| CFTR-related disorder | Two mild or heterozygous | 40–60 mmol/L | Single-organ disease (CBAVD, bronchiectasis, pancreatitis) |
Part 2 — CFTR Modulator Therapy (Disease-Modifying)
CFTR modulators address the root cause — defective CFTR protein. They represent the most transformative advance in CF treatment.
Mutation Classes and Modulator Types
| CFTR Mutation Class | Defect | Drug Class | Examples |
|---|---|---|---|
| Class I (nonsense) | No protein produced | Read-through agents | Investigational |
| Class II (misfolding) | Protein misfolded, not trafficked | Correctors | Lumacaftor, tezacaftor, elexacaftor |
| Class III (gating) | Protein reaches surface but won't open | Potentiators | Ivacaftor |
| Class IV/V (reduced function/quantity) | Reduced activity or expression | Potentiators | Ivacaftor |
Approved CFTR Modulators
| Drug (Brand) | Class | Mutations | Notes |
|---|---|---|---|
| Ivacaftor (Kalydeco) | Potentiator | G551D + 9 other gating mutations | Single-drug; highly effective for gating mutations |
| Lumacaftor/ivacaftor (Orkambi) | Corrector + potentiator | F508del homozygous | Modest efficacy; largely superseded |
| Tezacaftor/ivacaftor (Symdeko/Symkevi) | Corrector + potentiator | F508del homozygous; some residual-function mutations | Better tolerated than Orkambi |
| Elexacaftor/tezacaftor/ivacaftor (Trikafta/Kaftrio) | Triple therapy | F508del + minimal function; F508del homozygous; F508del + gating/residual | Transformative — approved age 2+; ~87% of CF patients eligible |
| Vanzacaftor/tezacaftor/deutivacaftor (Alyftrek) | Next-gen triple | F508del + ETI-eligible variants; 31 additional rare mutations | FDA approved Dec 2024; once-daily dosing; superior sweat Cl⁻ reduction vs ETI |
Elexacaftor/Tezacaftor/Ivacaftor (ETI / Trikafta) — The Standard of Care
- Improves FEV₁ by ~14% predicted; reduces pulmonary exacerbations by ~63%; dramatically improves quality of life, weight, and sweat chloride
- Now approved down to age 2 years
- Approved for any CFTR mutation producing a CFTR protein (expanded FDA approval, 2025)
- Monitoring required: liver function tests (hepatotoxicity, though rare); mental health (anxiety, depression, "mental fog" reported in a minority)
- Murray & Nadel's Textbook of Respiratory Medicine; Thompson & Thompson Genetics 9e
Vanzacaftor/Tezacaftor/Deutivacaftor (VTD / Alyftrek) — New in 2024
SKYLINE Phase III RCTs (Lancet Respir Med 2025 — PMID 39756424):
- 12 years+ with multiple genotypes (F508del-MF and other ETI-eligible)
- VTD non-inferior to ETI on FEV₁ at 24 weeks, with superior sweat chloride reduction (greater CFTR functional restoration)
- Once-daily dosing (vs ETI's twice-daily ivacaftor component)
- Approved Dec 2024 for ages 6+; also covers 31 additional rare mutations not previously eligible for modulators
Post-ETI Supportive Therapy Reduction
SIMPLIFY RCT (Ann Am Thorac Soc 2024 — PMID 39041864):
- Patients on ETI who discontinued both hypertonic saline and dornase alfa showed no meaningful change in FEV₁ or lung clearance index
- Significant reduction in treatment burden reported
- Key practice implication: many supportive inhaled therapies can be de-escalated in patients on highly effective modulators (HEMTs) — but should be done in shared decision-making
Mutations Without Modulator Options (Class I)
- ~10% of patients have class I (nonsense) mutations producing no CFTR protein — current modulators are ineffective
- Gene therapy, mRNA replacement, and CRISPR-Cas9 approaches are under active investigation but not yet approved
Part 3 — Pulmonary Management
Physical Airway Clearance (Daily — Standard of Care)
- Chest physiotherapy (manual vibropercussion, oscillating vest systems)
- Positive expiratory pressure (PEP) devices, flutter valves, active cycle of breathing
- Aerobic exercise (70–85% max HR) — improves mucus clearance and exercise tolerance
- Pulmonary rehabilitation for severe disease
Mucoactive / Airway Rehydration Therapies
| Agent | Mechanism | Notes |
|---|---|---|
| Dornase alfa (Pulmozyme) | Cleaves extracellular DNA in mucus → reduces viscosity | 2.5 mg inhaled once daily; improves FEV₁ ~6%, reduces exacerbations; may be reduced/stopped on HEMT (see above) |
| Hypertonic saline (7%) | Osmotic rehydration of airway surface liquid | 4 mL inhaled BID; reduces exacerbations 56%; may be de-escalated on HEMT |
| Mannitol (inhaled) | Osmotic agent | Alternative where hypertonic saline not tolerated |
Anti-Infective Therapy
Preventing/eradicating early Pseudomonas aeruginosa:
- Early detection and aggressive eradication is critical — chronic colonisation markedly accelerates pulmonary decline
- Inhaled tobramycin (TOBI) or aztreonam (Cayston) alternating monthly; or inhaled ciprofloxacin (dry powder)
- IV antibiotics (tobramycin + anti-pseudomonal β-lactam) for pulmonary exacerbations
Chronic P. aeruginosa management:
- Chronic suppressive inhaled antibiotics (tobramycin, aztreonam, colistin)
- IV course (2–3 weeks) for acute exacerbations, typically at a CF centre
Common pathogens by progression:
| Stage | Organisms |
|---|---|
| Early childhood | S. aureus (MRSA increasingly common), H. influenzae |
| Adolescence/adulthood | P. aeruginosa (80% by age 18) |
| Advanced disease | Burkholderia cepacia (very poor prognosis), NTM, ABPA |
Burkholderia cepacia: Nosocomial spread risk — strict patient segregation required; associated with "cepacia syndrome" (fatal necrotising pneumonia).
NTM (Mycobacterium abscessus, M. avium complex): Increasing prevalence in CF; prolonged multiagent regimens required; consultation with specialist.
ABPA (Allergic Bronchopulmonary Aspergillosis): Affects ~10% of CF patients; treat with oral corticosteroids ± itraconazole/voriconazole.
Anti-Inflammatory Therapy
- Azithromycin (250–500 mg, 3× weekly): reduces pulmonary exacerbations by ~40% through anti-inflammatory and anti-biofilm mechanisms; standard of care for patients with Pseudomonas colonisation
- Ibuprofen (high-dose): slows FEV₁ decline in children aged 5–13; under-utilised due to monitoring requirements
- Systemic corticosteroids: for ABPA; not routinely used for CF lung disease
Pulmonary Exacerbations
- Defined by worsening respiratory symptoms, ↓FEV₁, increased sputum
- Treated with IV antibiotics (typically 14–21 days), intensified airway clearance, nutritional support
- Goal: return FEV₁ to baseline — failure to recover predicts long-term decline
- Hospital or home IV therapy depending on severity
Lung Transplantation
- Indicated when FEV₁ <30% predicted, rapid decline, or refractory respiratory failure
- Bilateral sequential lung transplant is standard; median post-transplant survival ~6 years
- Listing criteria: 2-year survival on transplant >50%
- ETI can delay need for transplant significantly; not a contraindication to transplant
Part 4 — Gastrointestinal & Nutritional Management
Pancreatic Exocrine Insufficiency (85–90% of patients)
- Pancreatic enzyme replacement therapy (PERT): lipase 500–2500 units/kg/meal; dose titrated to stool consistency and weight gain
- Standard: enteric-coated microsphere preparations (Creon, Zenpep)
- Always taken with meals and snacks; do not crush
Nutritional Support
- CF is a hypermetabolic state; caloric needs are 120–150% of normal
- Target BMI ≥50th percentile (children), ≥22 kg/m² (adult women), ≥23 kg/m² (adult men)
- Fat-soluble vitamins (A, D, E, K) must be supplemented — malabsorption is universal in PI
- High-calorie diet with supplemental enteral feeds via gastrostomy if needed
- ETI markedly improves nutritional status and weight in most patients
- ESPEN-ESPGHAN-ECFS Nutrition Guideline 2024 (PMID 38169175)
Specific GI Complications
| Complication | Management |
|---|---|
| Meconium ileus (10–25% of newborns) | Gastrografin enema; surgery if failed |
| Distal intestinal obstruction syndrome (DIOS) | Oral Gastrografin or PEG; polyethylene glycol laxatives; hydration |
| Rectal prolapse | Manual reduction; improved nutrition and PERT |
| CF-related liver disease | Ursodeoxycholic acid (UDCA); monitor LFTs; portal hypertension management |
| CF-related diabetes (CFRD) | Insulin (first-line); oral hypoglycaemics generally not recommended; annual OGTT screening from age 10 |
| GERD | PPI; especially important before modulator therapy |
Part 5 — Monitoring & Multidisciplinary Management
Routine Monitoring Schedule
| Parameter | Frequency |
|---|---|
| Pulmonary function tests (spirometry) | Every visit (minimum quarterly) |
| Sputum culture and sensitivity | Every visit |
| Chest X-ray | Annually (CT chest when clinically indicated) |
| LFTs (especially on CFTR modulators) | Every 3 months initially, then 6-monthly |
| OGTT for CFRD screening | Annually from age 10 |
| Bone density (DXA) | Every 1–5 years from age 18 |
| Fat-soluble vitamin levels (A, D, E, K) | Annually |
| Nutritional assessment | Every visit |
Multidisciplinary CF Team
- Pulmonologist, gastroenterologist, dietitian, physiotherapist, CF specialist nurse, pharmacist, social worker, psychologist
- Segregation protocols: patients should not share waiting rooms or equipment — organisms can spread between patients (especially P. aeruginosa and B. cepacia)
Reproductive Considerations
- ~95% of males are infertile (CBAVD/obstructive azoospermia) — sperm retrieval for ICSI possible
- Women have reduced fertility but can conceive; pregnancy management requires specialist input
- Preconception genetic counselling; carrier screening for partners
Summary: What Has Changed (2024–2025)
| Area | Update |
|---|---|
| Vanzacaftor/tezacaftor/deutivacaftor (Alyftrek) | FDA approved Dec 2024; once-daily; superior sweat Cl⁻ reduction vs Trikafta; eligible age 6+ including 31 new rare mutations |
| ETI in younger children | Trikafta approved down to age 2 |
| De-escalation of inhaled therapies on HEMT | Discontinuing hypertonic saline + dornase alfa is safe in selected ETI patients (SIMPLIFY RCT, 2024) |
| Nutrition guideline update | ESPEN-ESPGHAN-ECFS 2024 guideline includes specific guidance for patients on highly effective modulators |
| Expanded mutation coverage | FDA 2025: ETI and Alyftrek approved for any protein-producing CFTR mutation (~800 additional patients eligible) |
Sources: Murray & Nadel's Textbook of Respiratory Medicine; Sleisenger & Fordtran's GI & Liver Disease; Harrison's Principles of Internal Medicine 22E; Thompson & Thompson Genetics & Genomics in Medicine 9e; Robbins Pathologic Basis of Disease; Goodman & Gilman's Pharmacological Basis of Therapeutics; SKYLINE Trials (Lancet Respir Med 2025, PMID 39756424); SIMPLIFY RCT (Ann Am Thorac Soc 2024, PMID 39041864); ESPEN-ESPGHAN-ECFS Nutrition Guideline (Clin Nutr 2024, PMID 38169175)
This is a shared conversation. Sign in to Orris to start your own chat.