Kartagener syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
Kartagener syndrome primary ciliary dyskinesia
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compose a thorough answer.
Kartagener Syndrome
Definition
Kartagener syndrome is a clinical subset of Primary Ciliary Dyskinesia (PCD) defined by the classic triad:
- Situs inversus (complete mirror-image reversal of visceral organs)
- Chronic sinusitis (and/or nasal polyposis)
- Bronchiectasis
It is also called the "Kartagener triad." When PCD is present without situs inversus, it is simply called PCD or immotile cilia syndrome. About 50% of PCD patients have situs inversus - those patients carry the Kartagener label.
- K.J. Lee's Essential Otolaryngology
- Scott-Brown's Otorhinolaryngology Head & Neck Surgery, Vol. 1
Genetics & Epidemiology
-
Inheritance: Autosomal recessive
-
Prevalence: ~1 in 20,000 births
-
Genetics: More than 45 genes are now known to cause PCD. Mutations affect genes encoding dynein arm proteins (e.g., DNAI1, DNAI2, DNAH5, DNAH11) and other structural components of cilia/flagella.
-
Men and women are equally affected in terms of respiratory disease.
-
Histology: A Text and Atlas, 9th ed.
-
The Harriet Lane Handbook, 23rd ed.
Pathophysiology
The core defect is a structural abnormality of cilia and flagella. In the normal cilium, movement is powered by dynein ATPase motors attached to the outer doublet microtubules (the "9+2" axoneme pattern: 9 peripheral doublets + 2 central microtubules). In Kartagener syndrome:
- Dynein arms are absent (both outer and inner) on the microtubule A-tubules
- Basal feet are misoriented, pointing in random directions rather than uniformly (seen on EM)
- The result is absent or severely impaired ciliary motility
This single molecular defect has wide consequences because motile cilia are present in many organ systems:
| Location | Normal role | Effect of defect |
|---|---|---|
| Respiratory epithelium | Mucociliary clearance | Mucus retention, recurrent infection |
| Paranasal sinuses | Sinus drainage | Chronic sinusitis, nasal polyps |
| Bronchi | Airway clearance | Bronchiectasis, recurrent pneumonia |
| Sperm flagellum | Motility | Male infertility (immotile sperm) |
| Female reproductive cilia | Ovum transport | Possible ectopic pregnancy |
| Embryonic nodal cilia | Left-right axis determination | Situs inversus in ~50% |
| Ependymal cilia | CSF circulation | Occasionally hydrocephalus |
- Histology: A Text and Atlas, 9th ed.
- Junqueira's Basic Histology, 17th ed.
Electron Micrograph - Absent Dynein Arms
Below is an EM cross-section of a cilium from a patient with PCD/Kartagener syndrome. Note the absence of dynein arms on the microtubule doublets (normally visible as small projections; here they are missing):
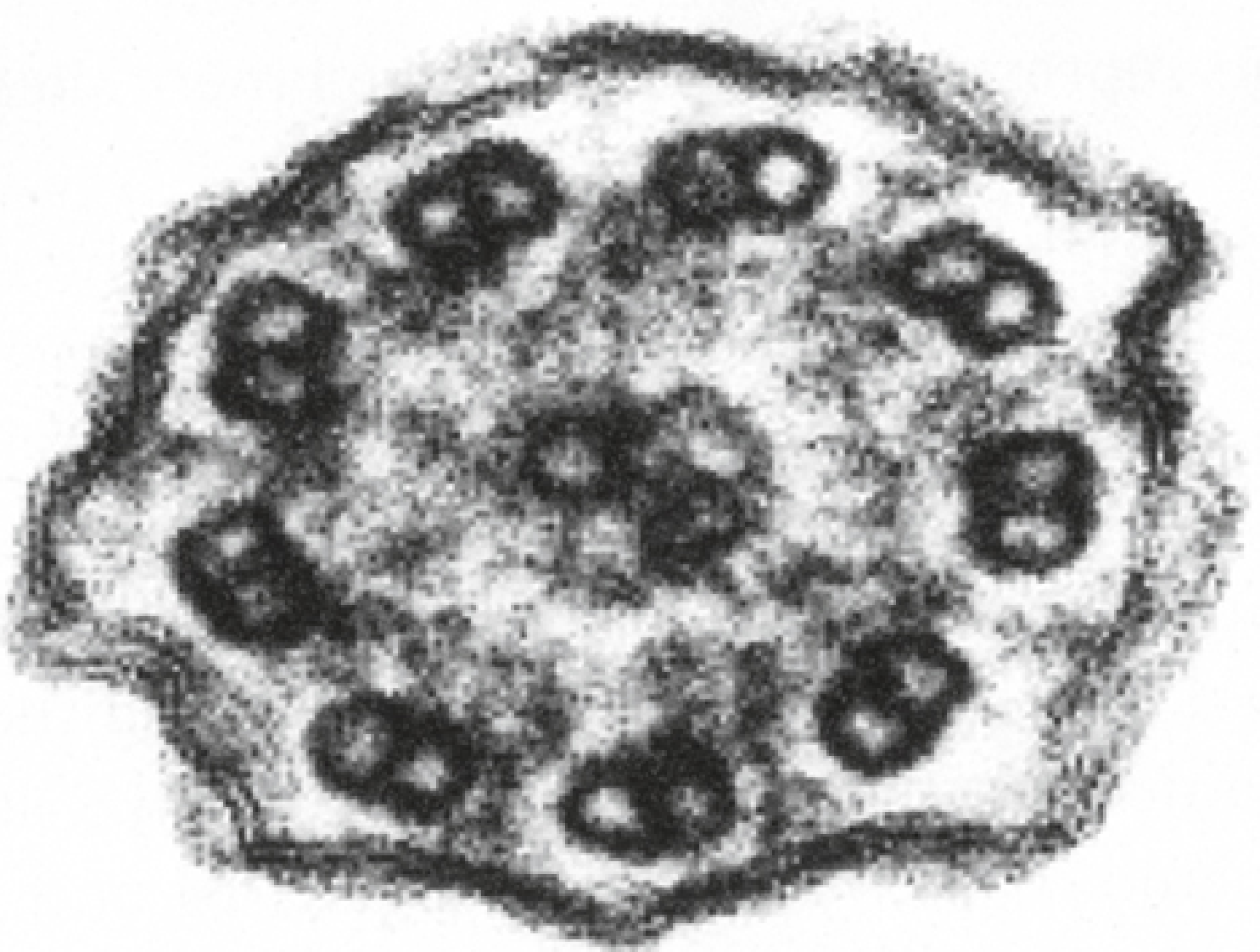
Electron micrograph (x180,000) showing absent dynein arms. - Histology: A Text and Atlas, 9th ed.
Clinical Features
Respiratory (most prominent):
- Recurrent lower respiratory tract infections since childhood
- Chronic productive cough
- Bilateral bronchiectasis - basal/lower-middle lobe predominance, usually cylindrical type
- Chronic rhinosinusitis
- Nasal polyposis (27% of Kartagener patients)
- Recurrent otitis media
- Asthma-like symptoms
Situs inversus:
- Complete visceral transposition (dextrocardia, liver on the left, etc.)
- Important clinical point: ECG leads and auscultation findings are mirror-image
- Seen on CXR as dextrocardia (heart apex pointing right)
Reproductive:
- Males: nearly complete or total infertility due to immotile but metabolically active sperm. Sperm motility <10% is a hallmark. Azoospermia is uncommon; the sperm are present but immotile.
- Females: potentially fertile, but increased risk of ectopic pregnancy due to impaired cilia transport in the Fallopian tubes
Neurological (rare):
-
Hydrocephalus or transient ventricular dilatation from impaired ependymal cilia
-
Campbell-Walsh-Wein Urology
-
Grainger & Allison's Diagnostic Radiology
-
Scott-Brown's Otorhinolaryngology
Chest X-Ray Findings
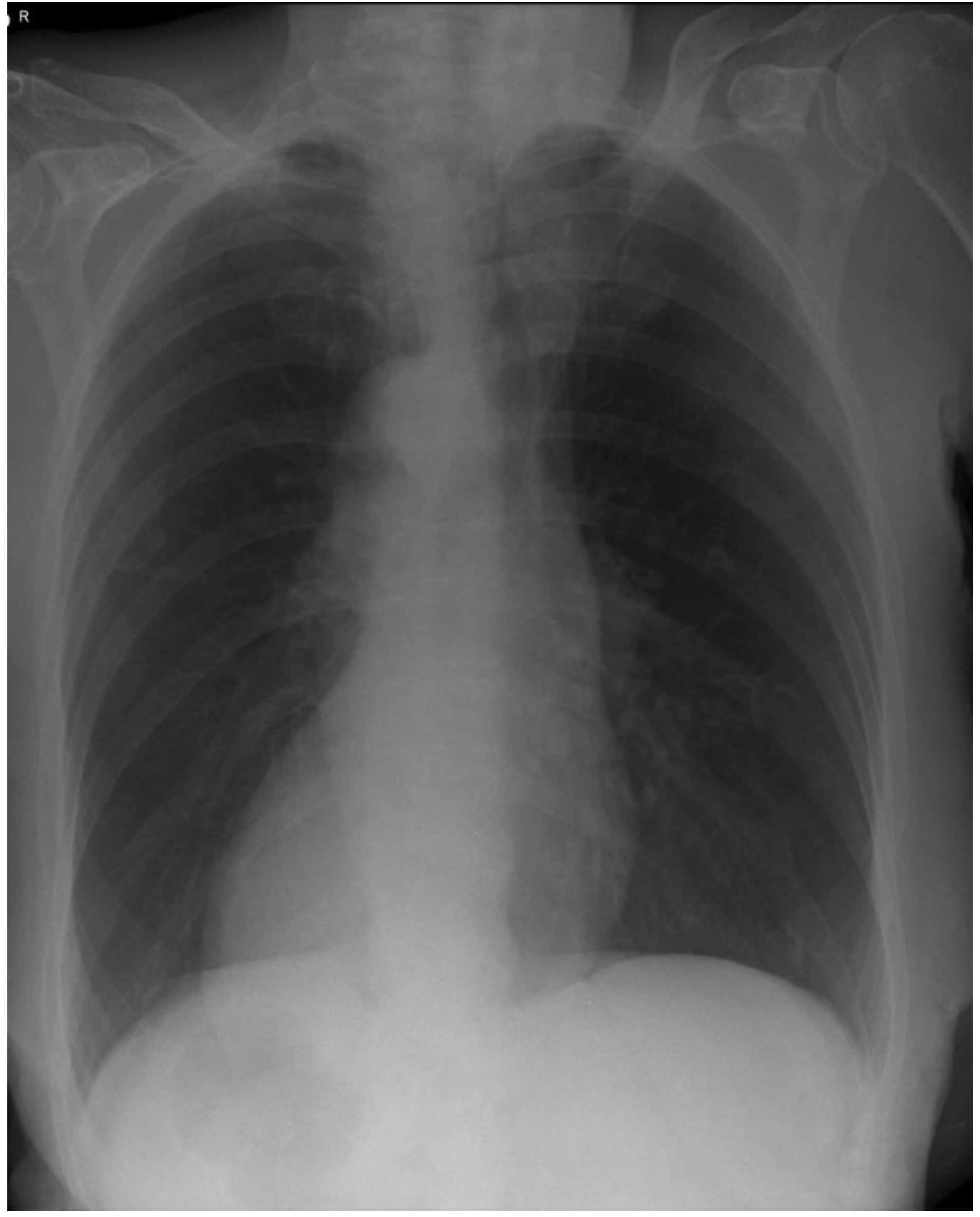
CXR showing dextrocardia (heart apex to the right). - Scott-Brown's Otorhinolaryngology
On imaging:
- CXR/CT: dextrocardia, situs inversus, bilateral bronchiectasis (lower > upper lobe), diffuse bronchiolitis
- CT sinuses: mucosal thickening, opacification of sinuses
Diagnosis
- Clinical suspicion: classic triad + childhood onset respiratory symptoms
- Nasal NO (nitric oxide): very low nasal NO is a sensitive screening test for PCD
- Electron microscopy (EM): gold standard - shows absent dynein arms and/or misoriented basal feet
- High-speed video microscopy: demonstrates abnormal or absent ciliary beat
- Genetic testing: >45 causative genes; broad gene panel testing recommended
- Semen analysis: <10% motility; EM of sperm tail may show structural defects
- Evaluation of other organ systems: abdominal US, echocardiogram, brain MRI
- The Harriet Lane Handbook, 23rd ed.
- Histology: A Text and Atlas, 9th ed.
Differential Diagnosis
Conditions that can mimic Kartagener/PCD:
-
Cystic fibrosis - also causes bronchiectasis, sinusitis, and infertility, but due to CFTR mutation (chloride channel), not ciliary defect
-
Common variable immunodeficiency - recurrent sinus and pulmonary infections, but no situs inversus or GI involvement
-
Young's syndrome - bronchiectasis + sinusitis + obstructive azoospermia, but normal ciliary ultrastructure (radial spoke/dynein malformation)
-
The Washington Manual of Medical Therapeutics
-
Scott-Brown's Otorhinolaryngology
Management
There is no curative treatment. Management is supportive and aimed at controlling respiratory disease:
- Airway clearance: chest physiotherapy, postural drainage, oscillating positive expiratory pressure devices
- Antibiotics: prompt and aggressive treatment of respiratory infections; long-term prophylactic macrolides (azithromycin) are used in some centers
- Bronchodilators/inhaled corticosteroids for airway inflammation
- Sinus surgery (FESS): for refractory chronic sinusitis / nasal polyps
- Fertility: males with Kartagener syndrome may father children using intracytoplasmic sperm injection (ICSI), as the sperm are viable despite being immotile
- Genetic counseling: autosomal recessive - 25% recurrence risk for siblings
- Regular surveillance: spirometry, sputum cultures, CT chest
Key Points Summary
| Feature | Detail |
|---|---|
| Triad | Situs inversus + chronic sinusitis + bronchiectasis |
| Inheritance | Autosomal recessive |
| Defect | Absent dynein arms on cilia/flagella (9+2 axoneme) |
| Key mechanism | Impaired mucociliary clearance |
| Male fertility | Infertile (immotile, metabolically active sperm) |
| Female fertility | Usually fertile, increased ectopic pregnancy risk |
| Diagnosis | Low nasal NO + EM showing absent dynein arms |
| Relationship to PCD | Kartagener = PCD + situs inversus (~50% of PCD cases) |
Recent literature note: Reviews published in Presse Med (2023, PMID 37516247) and Pediatrics (2024, PMID 38695103) provide updated guidance on PCD/Kartagener diagnosis and management, including emerging data on genetic heterogeneity and novel diagnostic criteria - these do not contradict the above but emphasize that molecular genetic testing is increasingly replacing EM as the primary diagnostic tool.
Fabrys disease management
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Fabry disease"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
I now have all the key information I need. Let me compile the comprehensive management answer.
Fabry Disease - Management
Background (Brief)
Fabry disease (Anderson-Fabry disease) is an X-linked lysosomal storage disorder caused by deficiency of α-galactosidase A (α-Gal A), encoded by the GLA gene on the X chromosome. The enzyme deficiency leads to progressive intracellular accumulation of globotriaosylceramide (Gb3) in vascular endothelial cells of the kidneys, heart, brain, and skin.
Classic hallmarks: acroparesthesias, angiokeratomas, anhidrosis, corneal opacities, and eventually renal failure, cardiomyopathy, and stroke.
Management Overview
Management of Fabry disease has two pillars:
- Disease-specific therapy - enzyme replacement therapy (ERT) or pharmacological chaperone therapy
- Organ-specific / symptomatic supportive therapy
1. Disease-Specific Therapies
A. Enzyme Replacement Therapy (ERT)
ERT has been available since 2001 and is the cornerstone of treatment.
| Drug | Brand | Dose | Source | Approval |
|---|---|---|---|---|
| Agalsidase alfa | Replagal (Takeda) | 0.2 mg/kg IV every 2 weeks (40 min infusion) | Human fibroblasts | EU, many countries (not US) |
| Agalsidase beta | Fabrazyme (Sanofi Genzyme) | 1.0 mg/kg IV every 2 weeks (several hours) | CHO cells | US (FDA-approved) + EU |
Who should receive ERT?
- All adult males with confirmed Fabry disease
- Symptomatic boys
- Symptomatic heterozygous females
- Initiated before irreversible end-organ damage for maximum benefit
Clinical effects:
- Clears microvascular endothelial Gb3 deposits from target organs
- Slows progression of renal, cardiac, and cerebrovascular complications
- Improves neuropathic pain, quality of life, GI symptoms, hypohidrosis, and autonomic function
- Better outcomes when started early - higher baseline proteinuria, worse initial GFR, and delayed therapy are strong predictors of renal progression despite ERT
- Mean annual GFR decline on agalsidase alfa: -2.2 mL/min/1.73m² in men, -0.7 in women (FOS registry data)
Side effects:
-
Infusion-related reactions: fever, rigors, chills, flushing, chest discomfort, rash, pruritus, nasal congestion (>50% during first months)
-
IgG or IgE antibodies may develop - can inhibit ERT efficacy
-
Management of reactions: reduce infusion rate, antihistamines, paracetamol/acetaminophen, corticosteroids; pre-medicate if needed
-
ERT can be administered during dialysis sessions
-
National Kidney Foundation Primer on Kidney Diseases, 8th ed.
-
Bradley & Daroff's Neurology in Clinical Practice
B. Pharmacological Chaperone Therapy - Migalastat
Migalastat (Galafold, Amicus Therapeutics) is an oral pharmacological chaperone approved for Fabry disease.
| Feature | Detail |
|---|---|
| Mechanism | Binds and stabilizes misfolded but functional α-Gal A mutant enzyme in the ER, facilitating proper trafficking to lysosomes |
| Dose | 123 mg orally every other day |
| Route | Oral (major advantage over IV ERT) |
| Eligibility | Only for patients with amenable (responsive) GLA mutations - not all variants respond |
| Approval | EU and US (FDA 2018) |
Efficacy:
- Phase III ATTRACT trial (18-month, randomized): migalastat was non-inferior to ERT in eGFR decline and superior in reducing LV mass index
- FAMOUS observational study confirms real-world efficacy
- Reduces plasma lyso-Gb3 (treatment monitoring biomarker)
Key point: Genetic amenability testing is mandatory before prescribing migalastat - approximately 35-50% of Fabry patients carry amenable mutations.
- National Kidney Foundation Primer on Kidney Diseases, 8th ed.
- Goldman-Cecil Medicine
C. Emerging / Investigational Therapies
| Approach | Examples | Status |
|---|---|---|
| Next-generation ERT | Pegunigalsidase alfa (PRX-102) - PEGylated, longer half-life, better tissue penetration | Approved in some regions |
| Substrate reduction therapy | Lucerastat, venglustat - inhibit Gb3 synthesis | Clinical trials |
| Gene therapy | Lentiviral and AAV vectors for GLA gene delivery | Phase I/II trials |
| mRNA therapy | Deliver mRNA encoding α-Gal A | Early development |
- National Kidney Foundation Primer on Kidney Diseases, 8th ed.
2. Organ-Specific Supportive Management
Pain (Acroparesthesias)
Most debilitating early symptom, often from childhood.
| Situation | Treatment |
|---|---|
| Acute painful crises | Avoid triggers (rapid temperature changes, fever, exertion, hot environments); NSAIDs |
| Chronic neuropathic pain | Anticonvulsants: carbamazepine, phenytoin, gabapentin, pregabalin |
| Small-fiber neuropathy | ERT improves autonomic function; adjunctive analgesics |
- Bradley & Daroff's Neurology in Clinical Practice
- National Kidney Foundation Primer on Kidney Diseases, 8th ed.
Kidney Disease
| Stage | Management |
|---|---|
| Proteinuria / early CKD | ACE inhibitors or ARBs - reduce proteinuria and slow GFR decline; note: α-Gal A may inhibit endogenous ACE, causing BP drop during infusion |
| Advanced CKD / ESKD | Dialysis or kidney transplantation |
| Transplantation | Preferred over dialysis - Fabry nephropathy does NOT recur in the allograft; better outcomes than dialysis |
| Monitoring | Annual eGFR, urine protein/creatinine, urine Gb3, plasma lyso-Gb3 |
Note: Dialysis patients with Fabry disease have poorer 3-year survival vs. non-diabetic controls. Kidney transplant 5-year survival is also lower than general controls but significantly better than dialysis.
- Comprehensive Clinical Nephrology, 7th ed.
- National Kidney Foundation Primer on Kidney Diseases, 8th ed.
Cardiac Management
Fabry cardiomyopathy is characterized by progressive LV hypertrophy, short PR interval, and eventual conduction disease.
| Manifestation | Treatment |
|---|---|
| LV hypertrophy / cardiomyopathy | ERT (slows progression); avoid pure vasodilators - may worsen outflow obstruction |
| Arrhythmias / conduction disease | Antiarrhythmics as indicated; pacemaker for significant bradycardia or heart block |
| Angina (small-vessel disease) | Antianginals; note: epicardial coronary disease is uncommon |
| Atrial fibrillation | Rate/rhythm control, anticoagulation |
| Heart failure | Standard HF therapies; ERT |
| Valvular disease | Surgical repair/replacement if severe |
- Braunwald's Heart Disease, 11th ed.
- Eur Heart J 2024 (PMID 38486361)
Cerebrovascular Disease
Stroke is seen in up to 25% of classic Fabry patients (predominantly posterior circulation).
| Measure | Approach |
|---|---|
| Secondary stroke prevention | Antiplatelet agents (aspirin, clopidogrel); anticoagulation if AF present |
| Hypertension | Aggressive BP control |
| Monitoring | Annual brain MRI in patients with neurological symptoms; white matter lesions on T2/FLAIR |
Other Supportive Treatments
| Symptom/Complication | Management |
|---|---|
| Hypohidrosis | Avoid hot environments; cooling strategies; temperature regulation |
| Angiokeratomas | Cosmetic removal with argon laser therapy |
| GI symptoms (nausea, bloating, diarrhea) | Dietary modification; prokinetics; small frequent meals; metoclopramide |
| Hearing loss | Hearing aids; prompt audiological evaluation |
| Ocular (cornea verticillata, cataracts) | Slit-lamp monitoring; specialist referral |
| Autonomic dysfunction | Compression stockings for orthostatic hypotension; fludrocortisone |
- National Kidney Foundation Primer on Kidney Diseases, 8th ed.
- Scott-Brown's / Bradley & Daroff's
3. Monitoring During Treatment
- Plasma lyso-Gb3: primary biomarker for treatment response and disease activity
- Urine Gb3: elevated early, decreases with ERT
- eGFR + urine protein: annual minimum
- Echocardiogram + ECG: annual (LV mass, wall thickness, valves, conduction)
- Brain MRI: every 2-3 years, or as indicated
- Anti-α-Gal A antibodies: check if poor response to ERT; antibody-mediated inhibition of ERT is a recognized mechanism of treatment failure
4. Diagnosis Essentials (Before Starting Treatment)
| Step | Details |
|---|---|
| Males | Measure leukocyte α-Gal A enzyme activity; if reduced, confirm with GLA sequencing |
| Females | Genetic testing mandatory - enzyme activity may be normal in carriers |
| Mutation amenability | Test GLA variant for migalastat amenability before prescribing chaperone therapy |
| Kidney biopsy | Useful at baseline to assess degree of fibrosis and stage nephropathy |
| Screening | Family cascade screening mandatory; screen all first-degree relatives |
5. Treatment Decision Algorithm (Summary)
Confirmed Fabry disease
|
├── Amenable GLA mutation?
│ YES → Migalastat 123 mg oral every other day
│ NO → ERT (agalsidase beta 1 mg/kg IV q2w [US]
│ or agalsidase alfa 0.2 mg/kg IV q2w [EU])
│
├── Kidney failure → Transplant preferred over dialysis
│
├── Pain → Anticonvulsants (carbamazepine/pregabalin)
│
├── Cardiac → ERT + standard HF/arrhythmia management
│
└── Stroke prevention → Antiplatelets ± anticoagulation
Recent evidence flag: A 2025 systematic review on Fabry cardiomyopathy (PMID 40840785, Prog Cardiovasc Dis) and a 2025 review on progress in Fabry treatment (PMID 40310476, BioDrugs) highlight that despite ERT, disease progression continues in a significant proportion of patients, strengthening the case for early initiation and next-generation therapies including gene therapy currently in trials. A 2024 European Heart Journal review (PMID 38486361) provides updated cardiologist-focused management guidance.
This is a shared conversation. Sign in to Orris to start your own chat.