Myocardial infraction
Reading File
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND "management"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive information to write a thorough answer. Let me compile everything.
Myocardial Infarction (MI)
Myocardial infarction - commonly called a heart attack - is death of cardiac muscle due to prolonged ischemia. Roughly 800,000 individuals in the United States experience an MI each year (nearly one every 40 seconds), causing almost 400,000 deaths annually. The major underlying cause is atherosclerosis.
Definition & Classification
| Type | Mechanism | ECG Pattern |
|---|---|---|
| STEMI (ST-elevation MI) | Complete transmural occlusion | ST elevation, Q waves |
| NSTEMI (Non-ST-elevation MI) | Partial/subendocardial occlusion | ST depression or T-wave changes |
Epidemiology
- 10% of MIs occur in people younger than 40 years; 45% in people younger than 65 years
- Male sex increases relative risk through middle age; females are generally protected during reproductive years
- Postmenopausal decline in estrogen leads to worsening coronary artery disease (CAD) - IHD is the most common cause of death in older females
- Post-menopausal hormonal replacement therapy has not been shown to be protective
(Robbins & Cotran Pathologic Basis of Disease)
Pathogenesis
Coronary Arterial Occlusion - The Classic Sequence
- An atheromatous plaque is disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to blood
- Platelets adhere, aggregate, and activate - releasing thromboxane A2, ADP, and serotonin - causing further platelet aggregation and vasospasm
- Coagulation cascade activates via tissue factor, adding to the growing thrombus
- Within minutes, the thrombus completely occludes the coronary artery lumen
When angiography is performed within 4 hours of MI onset, thrombotic occlusion is seen in almost 90% of cases. By 12-24 hours, only 60% show thrombosis - because some occlusions clear spontaneously.
Non-Atherosclerotic Causes (~10% of cases)
- Vasospasm (with or without atherosclerosis) - e.g., cocaine, ephedrine
- Embolism (from mural thrombus in atrial fibrillation, infective endocarditis, prosthetic valves, patent foramen ovale)
- Vasculitis
- Hematologic abnormalities (e.g., sickle cell disease)
- Amyloid deposition in vessel walls
(Robbins & Cotran Pathologic Basis of Disease)
Myocardial Response to Ischemia
| Event | Time |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | < 2 minutes |
| ATP reduced to 50% of normal | 10 minutes |
| ATP reduced to 10% of normal | 40 minutes |
| Irreversible cell injury (necrosis) | 20-40 minutes |
| Microvascular injury | > 1 hour |
- The subendocardial zone is most susceptible - it is the last area to receive blood from epicardial vessels and is exposed to high intramural pressures
- With prolonged ischemia, a "wavefront" of necrosis moves centripetally from subendocardium toward epicardium
- Benefits of reperfusion are greatest when achieved quickly - this is the rationale for rapid diagnosis and early intervention
(Robbins & Cotran Pathologic Basis of Disease)
Types of Infarction (by Location)
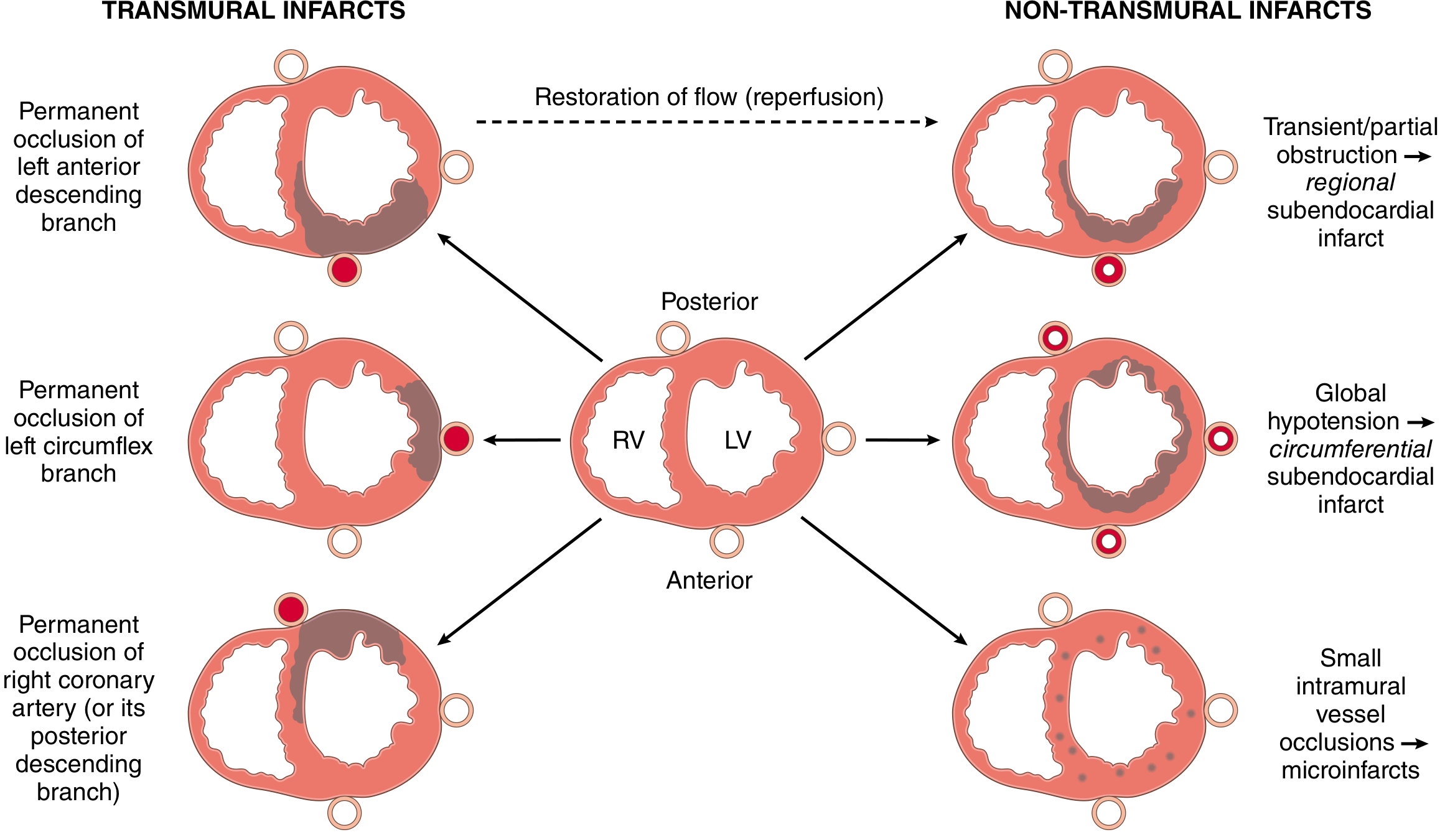
| Type | Cause | Pattern |
|---|---|---|
| Transmural | Permanent occlusion of epicardial artery | Full thickness of ventricular wall in distribution of occluded artery |
| Subendocardial | Plaque disruption with subsequent spontaneous/therapeutic lysis before full-thickness necrosis | Regional (one artery territory) |
| Circumferential subendocardial | Global hypotension superimposed on chronic stenoses | Circumferential, not limited to one artery |
| Microinfarction | Small intramural vessel pathology (vasculitis, microembolism, catecholamine excess) | Multifocal, scattered |
- LAD occlusion - anterior and septal wall infarction (most common and most lethal)
- RCA occlusion - posterior/inferior wall, often involves the right ventricle
- LCX occlusion - lateral wall of the left ventricle
Morphological Evolution
| Time | Gross Features | Light Microscopy | Electron Microscopy |
|---|---|---|---|
| 0-0.5 hr | None | None | Relaxation of myofibrils; glycogen loss; mitochondrial swelling |
| 0.5-4 hr | None | Usually none; variable waviness of fibers at border | Sarcolemmal disruption; mitochondrial amorphous densities |
| 4-12 hr | Dark mottling (occasional) | Early coagulative necrosis; edema; hemorrhage | - |
| 12-24 hr | Dark mottling | Coagulative necrosis; pyknotic nuclei; myocyte hypereosinophilia; marginal contraction band necrosis; early neutrophilic infiltrate | - |
| 1-3 days | Mottling with yellow-tan infarct center | Coagulative necrosis with loss of nuclei and striations; brisk neutrophilic infiltrate | - |
| 3-7 days | Hyperemic border; central yellow-tan softening | Disintegrating dead myofibers; dying neutrophils; early macrophage phagocytosis at border; early granulation tissue | - |
| 7-10 days | Maximally yellow-tan and soft; depressed red-tan margins | Well-developed phagocytosis; granulation tissue at margins | - |
| 10-14 days | Red-gray depressed infarct borders | Well-established granulation tissue with new blood vessels and collagen deposition | - |
| 2-8 weeks | Gray-white scar, progressive from border to center | Increased collagen deposition; decreased cellularity | - |
| > 2 months | Dense white scar | Dense collagenous scar | - |
Key histological stain: Triphenyl tetrazolium chloride (TTC) - stains intact viable myocardium brick-red; infarcted tissue appears as an unstained pale zone (dehydrogenases leak from dead cells).
(Robbins & Cotran Pathologic Basis of Disease)
ECG Changes
Three major electrical abnormalities in acute MI (Ganong's Medical Physiology):
| Defect in Infarcted Cells | Current Flow | ECG Change in Leads over Infarct |
|---|---|---|
| Rapid repolarization (accelerated K+ channel opening) | Out of infarct | ST segment elevation |
| Decreased resting membrane potential (loss of intracellular K+) | Into infarct | TQ segment depression (manifested as ST elevation) |
| Delayed depolarization | Out of infarct | ST segment elevation |
- Acute: ST elevation over the infarct, reciprocal ST depression in opposing leads
- After days/weeks: ST abnormalities subside; dead tissue is electrically silent
- Late: Pathological Q waves (negative relative to normal myocardium during systole); "failure of progression of the R wave" in anterior infarcts
- Non-Q-wave infarcts tend to be less severe but carry a high incidence of subsequent reinfarction
Biomarkers
| Biomarker | Rise | Peak | Duration |
|---|---|---|---|
| Cardiac Troponin I/T (cTnI/cTnT) | 2-4 hours | 24-48 hours | 7-10 days |
| CK-MB | 4-6 hours | 24 hours | 48-72 hours |
- Cardiac troponins are the most clinically useful and specific biomarkers
- Serial measurements help distinguish MI from other causes of troponin elevation (heart failure, pulmonary embolism, renal failure, sepsis - these do not follow the same time course)
- With reperfusion, troponin levels peak earlier and may be higher due to rapid washout from necrotic tissue
(Robbins & Cotran Pathologic Basis of Disease)
Treatment
Immediate Therapies (MONA + reperfusion)
- Oxygen - for patients with hypoxia (SpO2 < 90%) or respiratory distress
- Aspirin - antiplatelet
- Nitrates - vasodilation, relief of ischemic pain
- Morphine - pain control (use cautiously; may delay clopidogrel absorption)
- Beta-blockers - reduce myocardial oxygen demand
- Anticoagulation - heparin (UFH or LMWH)
- P2Y12 inhibitors - clopidogrel, ticagrelor, or prasugrel (dual antiplatelet therapy)
Reperfusion (the cornerstone of STEMI management)
Primary PCI (percutaneous coronary intervention) is the preferred strategy:
- Target: door-to-balloon time < 90 minutes
- Restores flow mechanically via balloon angioplasty + stent
- More effective than thrombolysis when performed in a timely manner at experienced centers
Thrombolysis (fibrinolytics - streptokinase, alteplase, tenecteplase):
- Used when PCI is not available within 120 minutes of first medical contact
- Target: door-to-needle time < 30 minutes
- Early thrombolysis and/or angioplasty can be highly successful in limiting myocardial necrosis
Long-term Secondary Prevention
- ACE inhibitors / ARBs - reduce remodeling, improve survival post-MI
- Statins - stabilize plaques, reduce LDL
- Beta-blockers - continued long-term
- Dual antiplatelet therapy - for 12 months (aspirin + P2Y12 inhibitor)
- Aldosterone antagonists (eplerenone) - in patients with reduced EF and heart failure post-MI
Complications
| Complication | Timing | Mechanism |
|---|---|---|
| Arrhythmias (VF, VT, heart block) | Minutes to hours | Electrical instability of ischemic myocardium |
| Cardiogenic shock | Hours to days | Pump failure (>40% LV necrosis) |
| Free wall rupture | 3-7 days (peak) | Softening of necrotic tissue before scar formation |
| Ventricular septal defect | 3-7 days | Septal rupture |
| Papillary muscle rupture | 3-7 days | Papillary muscle necrosis causing acute mitral regurgitation |
| Pericarditis (Dressler syndrome) | Weeks to months | Autoimmune reaction |
| Left ventricular aneurysm | Weeks | Expansion and fibrosis of infarcted segment |
| Mural thrombus with embolism | Days to weeks | Stasis over akinetic segment |
Sources:
- Robbins & Cotran Pathologic Basis of Disease - Myocardial Infarction chapter (pp. 510-517)
- Ganong's Review of Medical Physiology, 26th Ed. - ECG changes in acute MI (p. 534)
- Robbins & Kumar Basic Pathology - IHD and MI
This is a shared conversation. Sign in to Orris to start your own chat.